臨床特徵
- 神經鞘瘤 (schwannoma,neurilemmoma) 是常見的良性病灶,最常發生於第四、五十年,男女發生率相等;最常見於四肢(主要上肢),其次為頭頸部(含口腔、眼眶、唾液腺)。
- 兒童病例極罕見,先天性者更屬例外;多表現為單發、無痛、大小不一的皮下腫塊;偶可多發,此時罕與 von Recklinghausen 神經纖維瘤病相關。純皮內腫瘤少見。
- 也可發生於骨、胃腸道、胰、肝、腹膜後、縱膈、氣管、鼻咽、喉、甲狀腺、腎上腺、淋巴結等。神經症狀(疼痛、感覺異常)少見,除非為大型深部病灶;惡性轉變極罕見。單純切除後復發很少。
相關綜合徵與基因
- 第二型神經纖維瘤病 (Neurofibromatosis type II):NF2 基因位於 22q12.2,編碼 merlin 蛋白;以聽神經 schwannoma、皮膚腫瘤及其他中樞神經病變(meningioma、白內障、視網膜錯構瘤)為特徵。約 59% 患者有皮膚腫瘤,多為 schwannoma。Café-au-lait 斑可見於高達 33% 患者,但較第一型少。
- NIH 對 NFII 的診斷標準:雙側前庭 schwannoma 即可診斷;或有 NF2 一等親加上 30 歲前單側前庭 schwannoma 或兩項其他相關病變等。
- Schwannomatosis:多發皮膚 schwannoma(可伴脊髓與其他神經病灶),現視為獨立疾病;多為偶發,少數呈體染色體顯性遺傳。可能涉及 INI1 / SMARCB1 (22q11) 生殖系變異。
- 細胞遺傳學顯示 22q 物質喪失或 monosomy 22,對應 NF2 基因。
病理組織特徵
- 通常為圓形且必有被膜 (encapsulated),多位於皮下或更深處;典型呈 Antoni A 與 Antoni B 雙相型態。
- Antoni A 區:細胞較多,由緊密排列的梭形細胞構成,核呈漸尖、細長、波浪狀;核柵欄狀排列形成特徵性 Verocay bodies。可見間質膠原玻璃樣化與局部營養不良性鈣化。
- Antoni B 區:散在的梭形或星狀細胞置於疏鬆黏液樣間質中,常見慢性發炎細胞與壁玻璃樣化的小血管,可見微囊變化與含鐵血黃素沉積。
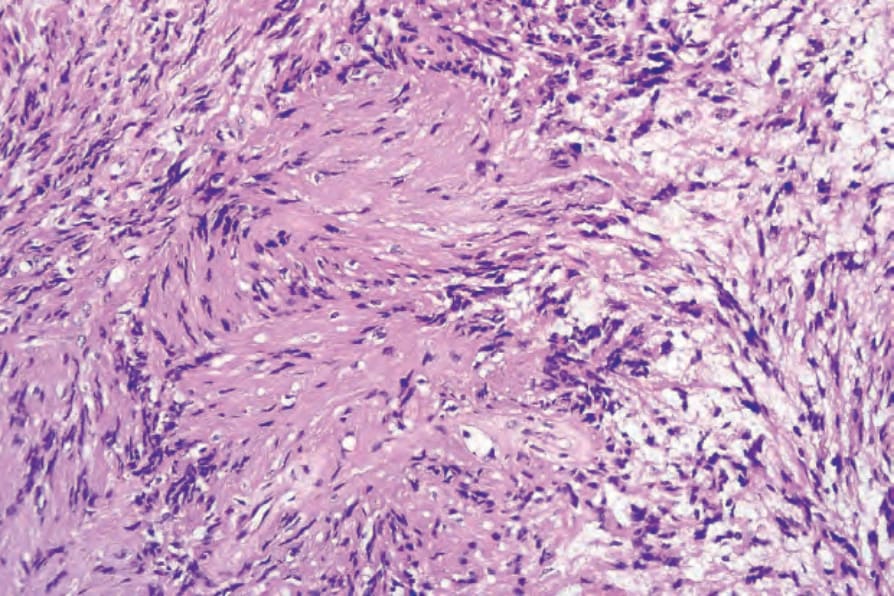
圖 35-311:Schwannoma,Antoni A 區典型的 Verocay body,由兩排平行核被 Schwann 細胞突起分隔。
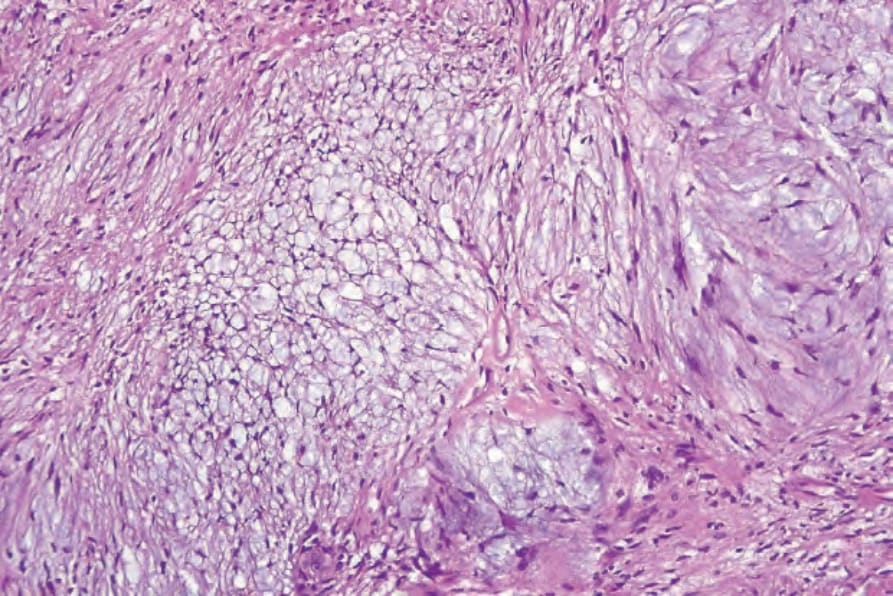
圖 35-312:Schwannoma,黏液樣退變形成 Antoni B 區。
免疫組化
- 超微結構上主要由 Schwann 細胞構成,反映為多數腫瘤細胞 S100 蛋白陽性。被膜含一層 EMA 陽性的神經周膜纖維母細胞。深部腫瘤可見 GFAP 與 CKAE1/AE3 陽性。多數病例 SOX10、podoplanin、calretinin 陽性;CD34 較局部。
- 腫瘤細胞表現 PDGFR-alpha、PDGFR-beta 配體及其受體與 KIT;imatinib mesylate 可抑制 schwannoma 細胞株。
變異型
- 叢狀 schwannoma (Plexiform):少與神經纖維瘤病相關(主要 NF2),多見於兒童或年輕成人頭頸部或軀幹;占所有 schwannoma 約 4.3%、皮膚 schwannoma 約 15%。多為小型,由 Antoni A 為主的多發被膜性結節構成。細胞型叢狀 schwannoma 見於嬰兒,可有 trisomy 17 復現,可能誤判為惡性,但無轉移潛能,局部復發不少見。
- 陳舊性 schwannoma (Ancient):多為深部、長期病灶,以顯著退變(核多形性、囊狀變化、鈣化、玻璃樣化、出血)為特徵,但有絲分裂少見。
- 細胞型 schwannoma (Cellular):細胞明顯增多並呈束狀結構,可類似平滑肌腫瘤;通常無 Verocay body。正常有絲分裂可達每 10 高倍視野 10 個,但無壞死或顯著核多形性;S100 陽性有助與平滑肌腫瘤區別。
- 惡性黑色素性 schwann 細胞腫瘤 (Malignant melanotic schwannian tumor):含色素細胞並常見 psammoma 小體;最常見於脊神經根周圍,皮膚表現極罕見(目前僅 20 例報告),其中 2 例轉移、1 例死於瀰漫性疾病;常與 Carney complex(黏液瘤、斑點色素沉著、內分泌過度活動;體染色體顯性,PRKAR1A 突變)相關。有絲分裂率大於每 10 HPF 2 個與轉移相關。
- 巴齊尼型 schwannoma (Pacinian):極罕見,多見於遠端肢體,由同心層狀的圓/卵形小體(似 pacinian 小體)構成。
- 上皮樣 / 神經母細胞瘤樣 schwannoma:罕見變異,多見於成人皮下或皮內;上皮樣細胞排列成索與巢,伴玻璃樣化或黏液樣間質;S100、SOX10 陽性,collagen type IV 勾勒個別單位或巢;約 42% 顯示 SMARCB1/INI1 喪失。行為良性,即使標為非典型者局部復發風險極低。
- 微囊 / 網狀 schwannoma:多見於胃腸道,部分發生於皮膚,呈微囊、網狀、蕾絲狀或假腺樣型態,背景含黏液樣物質;S100 瀰漫陽性。
雜交腫瘤與惡性轉變
- 兼具 schwannoma 與 neurofibroma 型態的雜交腫瘤日益被認識,常見於神經纖維瘤病患者,且 neurofibroma 多為叢狀型;在 schwannomatosis 與神經纖維瘤病患者中比例偏高。
- 惡性轉變常見多形性上皮樣細胞,罕見分化異向(如上皮樣血管肉瘤)。