Schwannoma
臨床特徵 (Clinical Features)
神經鞘瘤 (schwannoma,neurilemmoma) 為常見的良性病灶,最常發生於第四、第五個十年,兩性發生率相當,最常發生於四肢(主要為上肢),其次為頭頸部(包括口腔、眼眶與唾液腺)(Fig. 35.308)。1–5
兒童病灶非常罕見,先天性者更屬例外。6,7 它們最常表現為單發、無痛、大小不一的皮下腫塊(少見情況下可非常巨大),但偶爾可為多發,在此情境下少數與 von Recklinghausen neurofibromatosis 相關。8,9 偶可見明顯囊性變化。純真皮內的腫瘤罕見。10,11 皮膚病灶罕見呈聚集 (agminate) 排列,曾有一例與其上覆蓋的 anetoderma 相關。12,13 發生於陰莖與外陰的腫瘤極為罕見。14,15 部分腫瘤發生於其他部位,包括骨、胃腸道、胰臟、肝、後腹膜、縱膈、氣管、鼻咽、喉、甲狀腺、腎上腺與淋巴結。16–26 神經學症狀(包括疼痛與感覺異常 paresthesias)並不常見,除非為巨大、深部的病灶;惡性變化極為罕見(見 neurofibroma)。5,27,28 例外的案例包括一例可能與異物相關的皮膚病灶。29 單純切除後的復發非常少見。30
Neurofibromatosis type II(NF2 基因位於 22q12.2,編碼 merlin 蛋白)以聽神經鞘瘤 (acoustic schwannomas)、皮膚腫瘤及其他中樞神經系統病灶(包括 meningioma、cataract 與 retinal hamartoma)為特徵。31,32 約 59% 的患者有皮膚腫瘤,其中大多數為 schwannomas。32 患者僅少數會發展出 neurofibromas 或混合型病灶。Café-au-lait spots 可見於高達 33% 的患者,但這些往往較 neurofibromatosis type I 患者為少。32 Neurofibromatosis type II 中 neurofibromas 的發展,可能源於 neurofibromin 與 NF2 基因產物 merlin 在調控 RAS proto-oncogene 上的交互作用。33,34 皮膚 schwannomas 僅少數與 neurofibromatosis type II 相關。35
National Institute of Health (NIH) 對 NFII 的診斷標準如下:
• 雙側前庭神經鞘瘤 (Bilateral vestibular schwannomas),無需任何額外特徵即可診斷本病;
• 一等親罹患 NF2,且患者出現單側前庭神經鞘瘤且年齡小於 30 歲,或出現另兩種相關病灶(例如 glioma、meningioma、schwannoma、juvenile cortical cataract);
• 單側前庭神經鞘瘤合併任何其他兩種相關病灶,包括 glioma、meningioma、schwannoma、neurofibroma 或 juvenile cortical cataract;
• 多發性 meningiomas 合併上述任一病灶。23
多發性皮膚 schwannomas,伴或不伴脊神經及其他神經出現類似病灶者,已被稱為 schwannomatosis。雖然最初曾質疑它是否代表一個獨立的疾病實體,或僅為 neurofibromatosis type II 的變異型,但現已視為一個獨立的疾病。32,36–45 大多數案例似乎為散發性,但少數已被描述具有體染色體顯性遺傳模式。已有案例描述其與多發性 meningiomas 相關,以及一個具有惡性 rhabdoid tumors 易感性的家族。46–50 INI1 / SMARCB1 (22q11) 的生殖細胞系異常 (germline aberrations) 可能參與其中。
一種新的遺傳症候群已被描述,由多發性 schwannomas、多發性 nevi 與多發性 vaginal leiomyomas 組成。51 其 nevi 為先天性,但 schwannomas 與 vaginal leiomyomas 則於成年期發展。
近期一項研究發現,發展出單發性 schwannoma 或 meningioma 的兒童或年輕成人,通常具有遺傳易感性。52
致病機轉與組織學特徵 (Pathogenesis and Histologic Features)
Schwannomas 的細胞遺傳學研究顯示,不是 22q 物質的缺失,就是 monosomy 22,可能對應於編碼 neurofibromin 2 或 merlin 蛋白的 NF2 基因 (22q12.2)。34,35,53,54
Schwannomas 通常為圓形且一律具有包膜 (encapsulated),典型上見於皮下或更深部組織;原發性真皮內起源並不常見。純粹神經內 (intraneural) 的腫瘤屬例外。55 顯微鏡下,其特徵為典型的雙相 (biphasic) 模式,由細胞密集的 Antoni A 區與細胞稀疏的 Antoni B 區構成。
• Antoni A 區構成病灶中細胞較密集的成分,由相當緊密排列的梭形細胞組成,其細胞核呈漸尖、拉長、略呈波浪狀;核柵欄狀排列 (nuclear palisading) 為顯著特徵,產生特殊的 Verocay bodies (Figs 35.309–35.311)。這些
有時為主要特徵。56 Verocay 樣小體 (Verocay-like bodies) 可見於若干其他腫瘤,包括 dermatofibroma 與 leiomyoma。57 偶可見退化性核多形性 (degenerative nuclear pleomorphism) 與有絲分裂活性,但兩者往往在空間上互不相關。基質膠原的玻璃樣變 (hyalinization) 與局部營養不良性鈣化 (dystrophic calcification) 有時可見。
• Antoni B 區的典型特徵為不規則散布的梭形或星狀細胞,置於豐富而疏鬆的黏液樣 (myxoid) 基質中 (Fig. 35.312)。在這些區域內,散布的慢性發炎細胞與小血管(常具玻璃樣變的管壁)為顯著特徵 (Fig. 35.313)。局部退化性變化,包括微囊性變化與含鐵血黃素 (hemosiderin) 沉積,並不少見。曾有文獻記載一例帶有膠原球狀體 (collagenous spherulosis) 的 schwannoma,且其中一例出現腦膜上皮樣 (meningothelial-like) 漩渦結構。58,59 血管內 (intravascular) 表現屬例外。60
在良性 schwannomas 中出現明顯腺體分化 (glandular differentiation) 此一極罕見的發現,代表被包陷的正常附屬器結構之增生。61–64
雖然相對罕見,但兼具 schwannoma 與 neurofibroma 混合外觀的腫瘤日益受到認識。65–72 這些可見於 neurofibromatosis 患者,且其 neurofibroma 部分常為叢狀變異型 (plexiform variant)。73 在 schwannomatosis 與 neurofibromatosis 患者中,它們似乎佔比偏高。74 一例混合型案例顯示 monosomy 22(NF2 位點缺失),以及 CTNNA3 基因 (10q21.3) 的功能喪失突變 (loss of function mutations)。
Neurofibromatosis type II 中所見的 schwannomas 已被證實含有軸突 (axons)。76
惡性轉化 (Malignant transformation) 常顯示多形性上皮樣細胞,且少數出現分歧分化 (divergent differentiation),例如出現 epithelioid angiosarcoma。77–79
超微結構上,schwannomas 主要由 Schwann cells 組成,這在免疫組織化學上反映為大多數腫瘤細胞呈 S100 protein 陽性。包膜含有一層 EMA 陽性的 perineurial fibroblasts。GFAP 與 CKAE1/AE3 陽性主要可見於深部腫瘤。SOX10、podoplanin 與 calretinin 在大多數案例中為陽性,而 CD34 陽性則較為局灶性。80–85 腫瘤細胞表現 PDGFR-alpha、PDGFR-beta 配體及其同源受體,以及 KIT,且已證實 imatinib mesylate 可抑制一個 schwannoma 細胞株。86,87
變異型 (Variants88)
• 叢狀神經鞘瘤 (Plexiform schwannoma) 為不常見的腫瘤,僅極少數與 neurofibromatosis(主要為 NF2)相關,傾向主要發生於兒童或年輕成人的頭頸部或軀幹。89–98 這些病灶約佔所有 schwannomas 的 4.3%,約佔皮膚 schwannoma 的 15%。它通常為小型的真皮內或皮下病灶,特徵為多發性具包膜的結節,主要由 Antoni A 組織構成 (Fig. 35.314)。任何組織學型別的 schwannoma 皆可出現於叢狀腫瘤中,尤以細胞型變異型 (cellular variant) 為甚。細胞型叢狀神經鞘瘤 (Cellular plexiform schwannoma) 發生於嬰兒,並曾報告出現 trisomy 17 之復現。98 它表現出缺乏邊界局限或浸潤性生長模式、細胞密度增加,以及相對較高的有絲分裂活性。這些特徵可能導致惡性的診斷,尤其在小型切片中。它無轉移潛能,但局部復發並不罕見。89,98
• 一個叢狀與多結節性 schwannomas 的亞群侵犯主要的周邊神經。亦可發生深部腫瘤。可能出現核多形性(輕至中度)、有限的有絲分裂活性與局部壞死(後者見於深部病例),但復發並非其特徵,且無惡性潛能。與 plexiform neurofibroma 的區分至關重要,以避免不恰當的 von Recklinghausen disease(neurofibromatosis type 1)臨床診斷。少數情況下,腫瘤與 neurofibromatosis type 2 及 schwannomatosis 相關。
• 陳舊性神經鞘瘤 (Ancient schwannoma) 通常為位置較深、長期存在的病灶,特徵為明顯的退化性變化,表現為核多形性,伴隨廣泛的囊腫形成、鈣化、玻璃樣變或出血 (Figs 35.315 and 35.316)。99 然而,有絲分裂罕見。
• 細胞型神經鞘瘤 (Cellular schwannoma) 僅少數表現為皮下的大型具包膜腫塊。100-–102 顯微鏡下,細胞密度明顯增加,結合主要為束狀 (fascicular) 的結構,可能類似平滑肌腫瘤 (Figs 35.317 and 35.318)。一般不見 Verocay bodies。黃色瘤樣細胞 (Xanthomatous cells) 與淋巴球浸潤可能很明顯 (Fig. 35.319)。正常有絲分裂數目可達每 10 個高倍視野 10 個,但壞死與明顯核多形性皆非其特徵。藉由 S100 陽性可輕易與平滑肌腫瘤區分 (Fig. 35.320)。
• 惡性黑色素性許旺氏腫瘤 (Malignant melanotic schwannian tumor)(先前稱為 melanotic schwannoma、psammomatous melanotic schwannoma)為一罕見病灶,除了神經腫瘤的特徵外,含有色素細胞並通常顯示砂粒體 (psammoma bodies) (Fig. 35.321)。103,104 基於腫瘤會復發且可能轉移的觀察,近期已提議所有病灶應標示為 malignant melanotic schwannian tumor。它最常發生於脊神經根周圍,皮膚表現非常罕見,迄今僅有 20 例報告發生於真皮或皮下組織,其中兩例已發生轉移,一名患者死於瀰漫性疾病。105,106 已記載兩例與 nevus of Ota 相關發生的案例。107 此腫瘤通常與 Carney complex(myxomas、斑點狀色素沉著與內分泌過度活躍;體染色體顯性,伴 PRKAR1A 突變)相關 (Fig. 35.322)。108,109 與後者相關的病灶亦可能以轉移性疾病表現。110 腫瘤細胞對 S100 protein、SOX10 及其他黑色素細胞標記染色陽性。CD34 亦可能為陽性。111 例外情況下腫瘤可顯示叢狀模式。112 組織學特徵無法預測其行為,但有絲分裂率大於每 10 HPF 2 個者除外,此與轉移相關。103,113
• 環層小體性神經鞘瘤 (Pacinian schwannoma) 為非常罕見的腫瘤,表現為單發結節,最常見於遠端肢體。其組織學特徵為一具包膜的腫塊,由圓形或卵圓形、呈同心圓層狀排列的小體(略似 pacinian corpuscles)組成,置於膠原性梭形細胞基質中。114,115 雖然
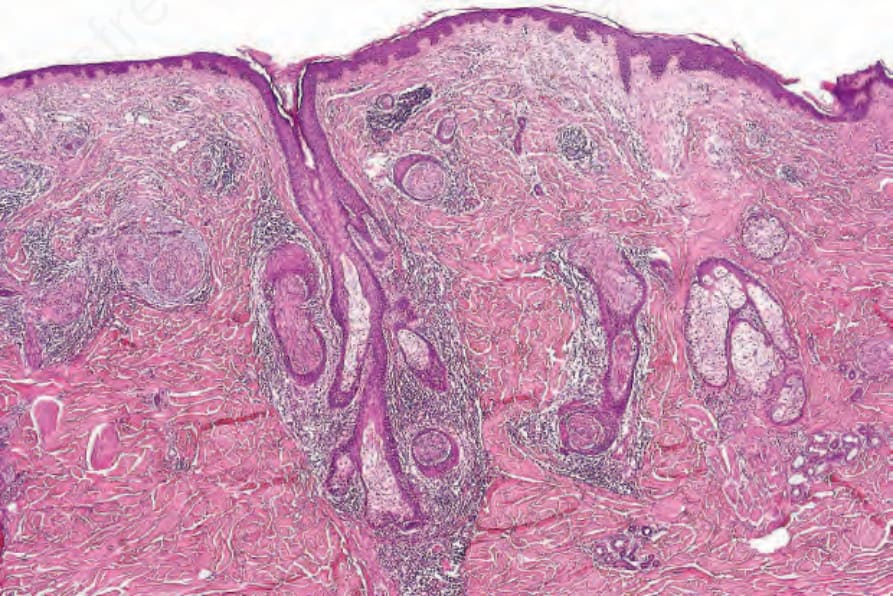
圖 35-306:上皮鞘神經瘤 (epithelial sheath neuroma):明顯的神經存在於淺層網狀真皮 (superficial reticular dermis),由溫和的鱗狀上皮巢 (nests of bland squamous epithelium) 所包裹。承蒙 L. Requena, MD, Madrid, Spain 提供。
Fig. 35.306 Epithelial sheath neuroma: prominent nerves are present in the superficial reticular dermis encased by nests of bland squamous epithelium. By courtesy of L. Requena, MD, Madrid, Spain.
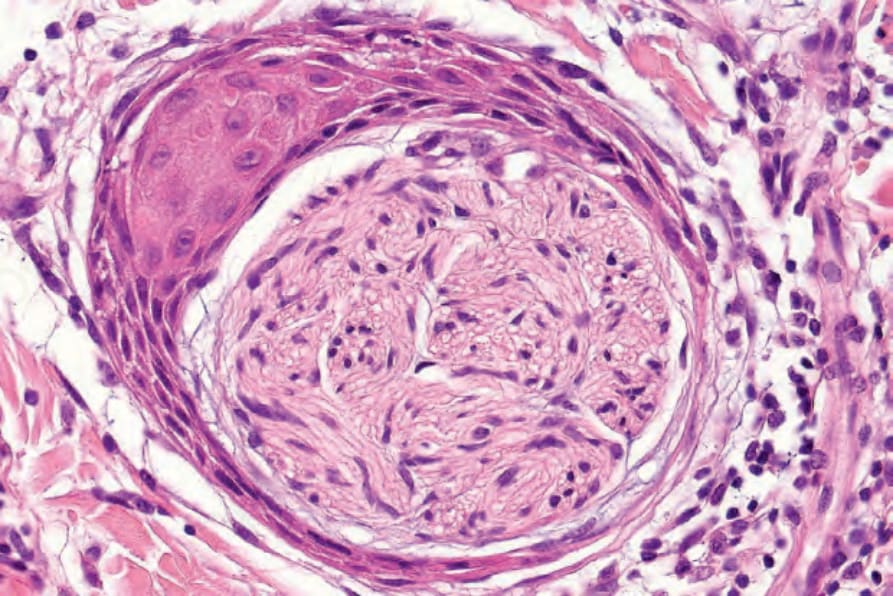
圖 35-307:上皮鞘神經瘤 (epithelial sheath neuroma):高倍視野。承蒙 L. Requena, MD, Madrid, Spain 提供。
Fig. 35.307 Epithelial sheath neuroma: high-power view. By courtesy of L. Requena, MD, Madrid, Spain.
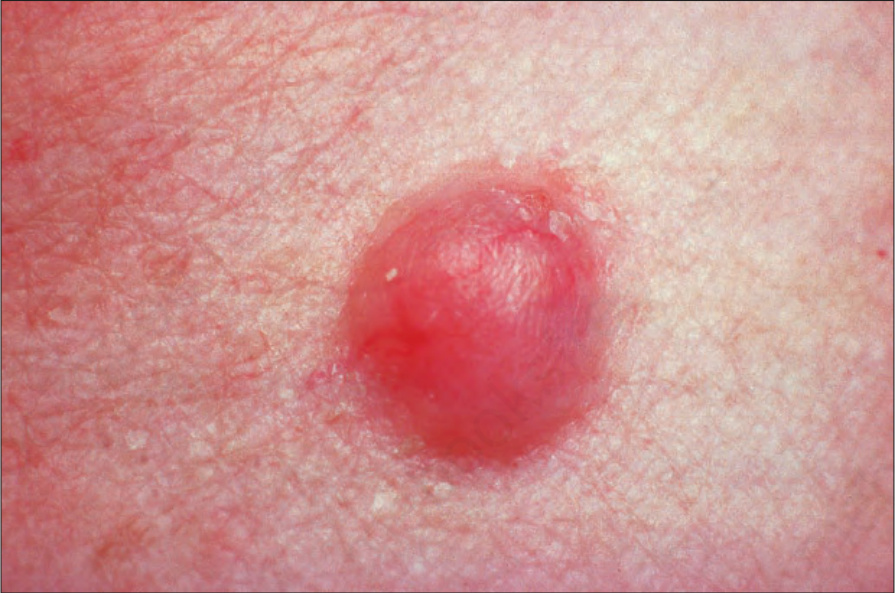
圖 35-308:神經鞘瘤 (schwannoma):此腫瘤表現為一非特異性的真皮結節 (non-specific dermal nodule)。承蒙 Institute of Dermatology, London, UK 提供。
Fig. 35.308 Schwannoma: this tumor presents as a non-specific dermal nodule. By courtesy of the Institute of Dermatology, London, UK.
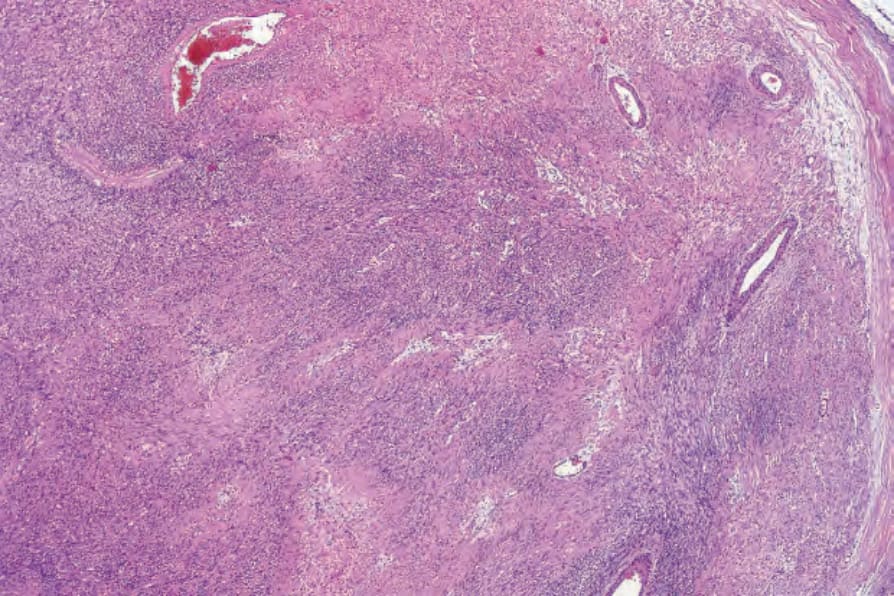
圖 35-309:神經鞘瘤 (schwannoma):梭形細胞腫瘤的掃描視野,具明顯的血管。右側可見包膜 (capsule)。
Fig. 35.309 Schwannoma: scanning view of spindle cell tumor with prominent blood vessels. A capsule is seen on the right side.
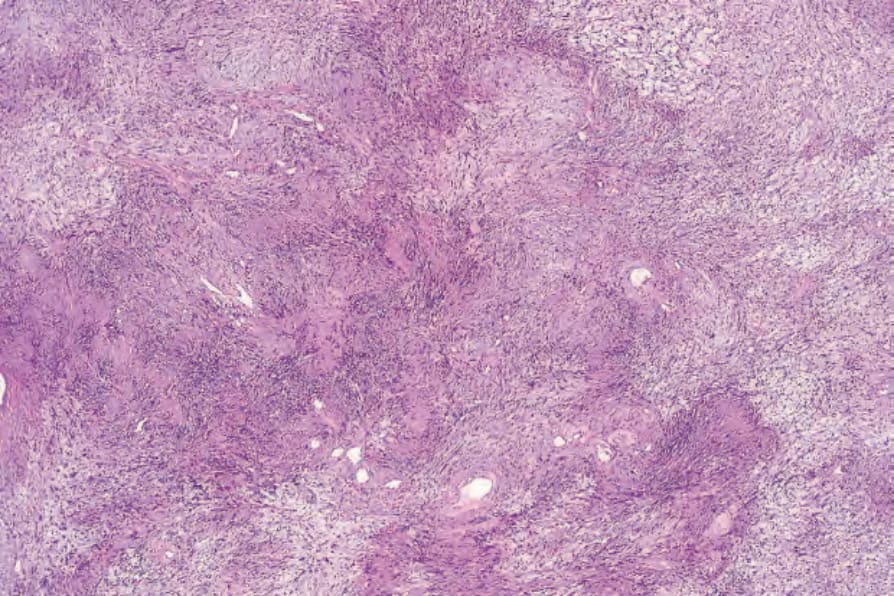
圖 35-310:神經鞘瘤 (schwannoma):柵欄狀排列 (palisading) 為其特徵性表現。
Fig. 35.310 Schwannoma: palisading is a characteristic feature.
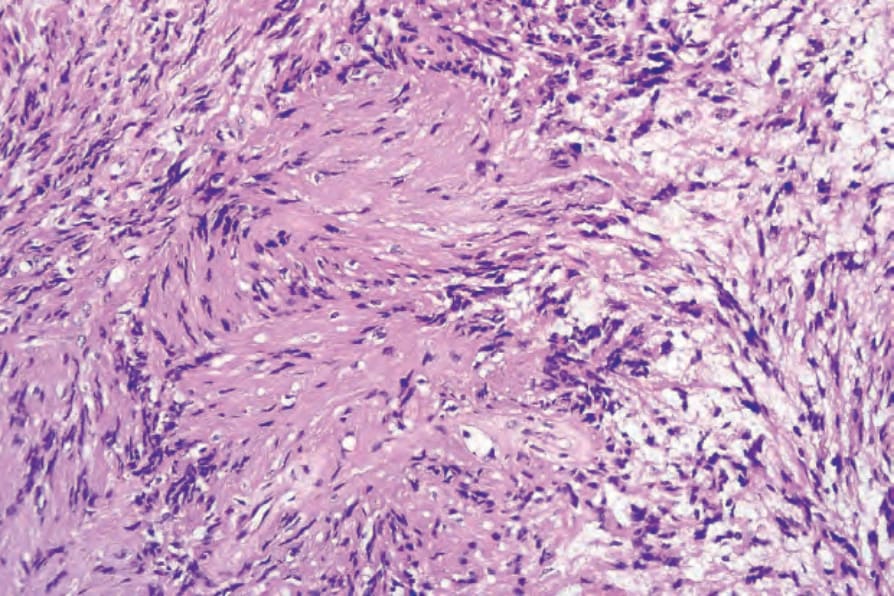
圖 35-311:神經鞘瘤 (schwannoma):Antoni A 區典型的 Verocay body,其特徵為兩排平行的核之間由 Schwann cell 突起 (Schwann cell processes) 分隔。
Fig. 35.311 Schwannoma: the Verocay body, typical of the Antoni A areas, is characterized by two parallel rows of nuclei separated by Schwann cell processes.
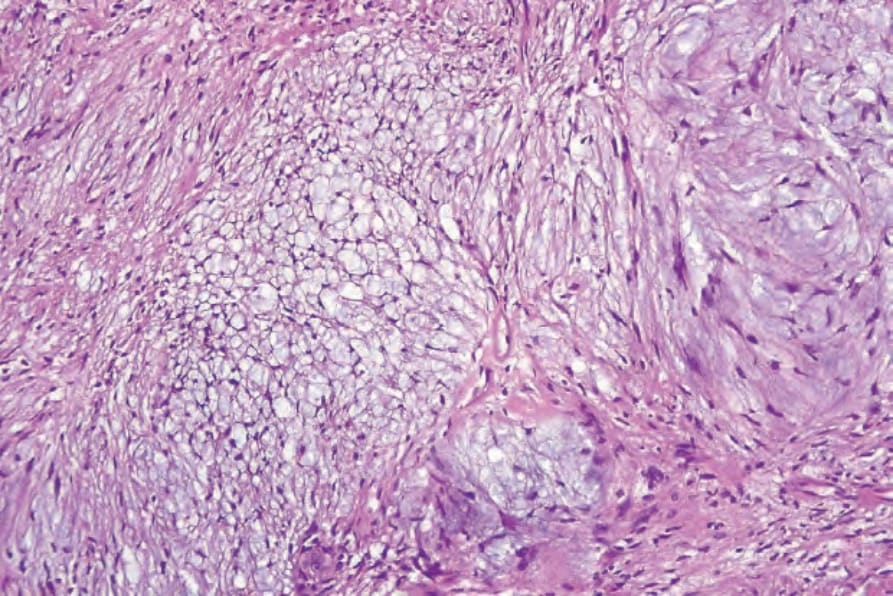
圖 35-312:神經鞘瘤 (schwannoma):黏液樣退化 (myxoid degeneration) 形成 Antoni B 區。
Fig. 35.312 Schwannoma: myxoid degeneration gives rise to Antoni B areas.
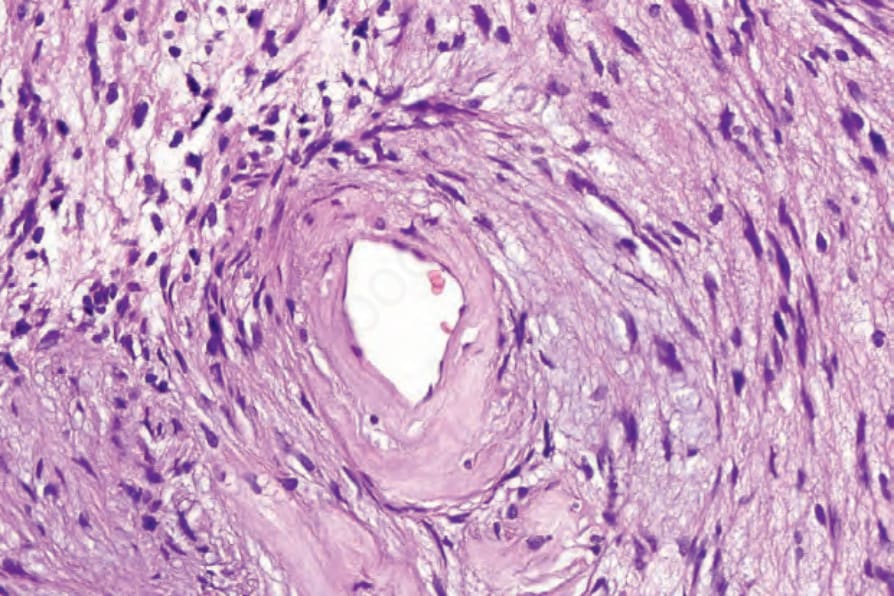
圖 35-313:神經鞘瘤 (schwannoma):Antoni B 區中血管壁有明顯的玻璃樣變 (hyalinization)。
Fig. 35.313 Schwannoma: there is marked hyalinization of the blood vessel walls in Antoni B.
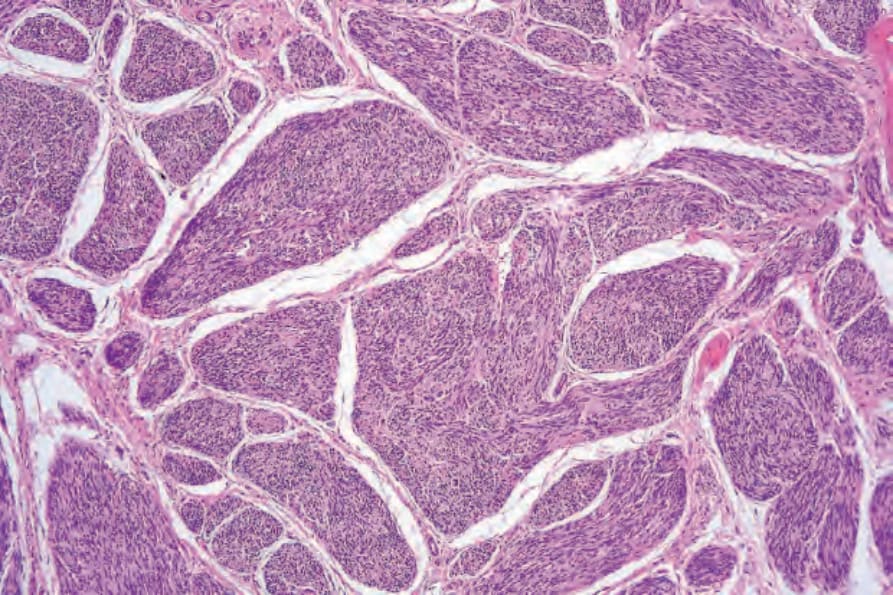
圖 35-314:叢狀神經鞘瘤 (plexiform schwannoma):此小型腫瘤由多個離散的神經鞘瘤性組織 (schwannomatous tissue) 結節組成。核柵欄狀排列 (nuclear palisading) 與 Verocay bodies 清晰可見。
Fig. 35.314 Plexiform schwannoma: this small tumor is composed of multiple discrete nodules of schwannomatous tissue. Nuclear palisading and Verocay bodies are evident.
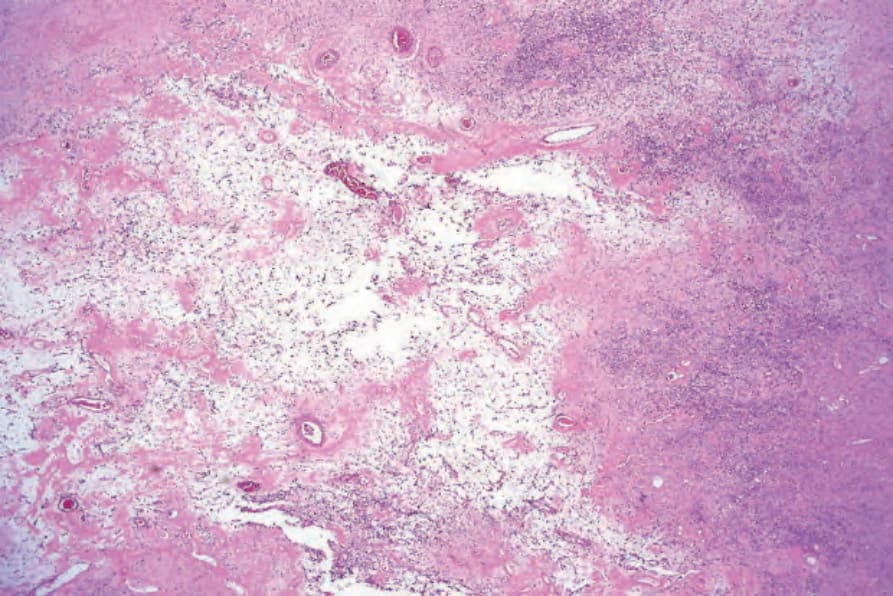
圖 35-315:陳舊性神經鞘瘤 (ancient schwannoma):退化性變化導致明顯的黏液樣特徵 (myxoid features),伴纖維化與顯著的血管增生。
Fig. 35.315 Ancient schwannoma: degenerative changes have resulted in marked myxoid features with fibrosis and conspicuous vascularity.
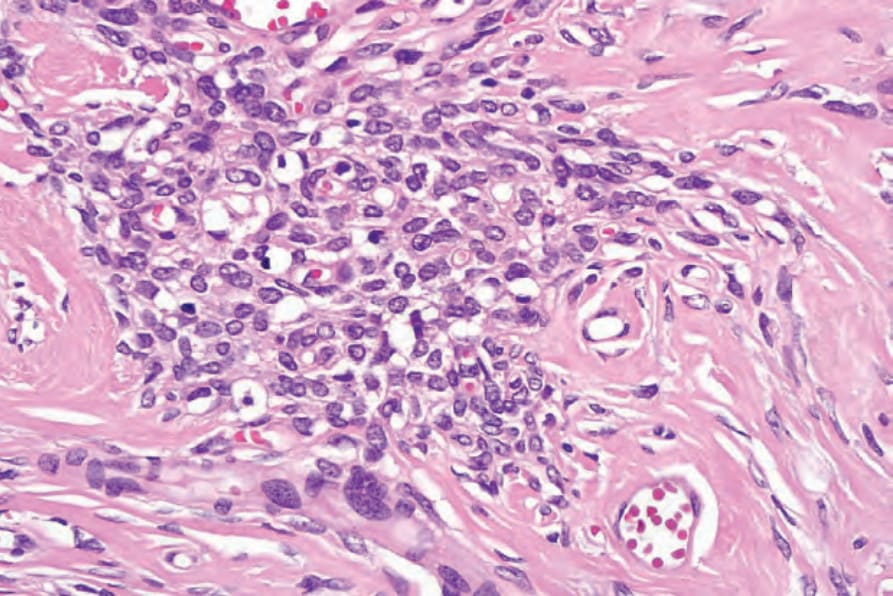
圖 35-316:陳舊性神經鞘瘤 (ancient schwannoma):局部核多形性 (focal nuclear pleomorphism) 不應被視為具有惡性意涵。這些腫瘤中不見有絲分裂活性。
Fig. 35.316 Ancient schwannoma: focal nuclear pleomorphism should not be taken as having sinister implication. Mitotic activity is not present in these tumors.
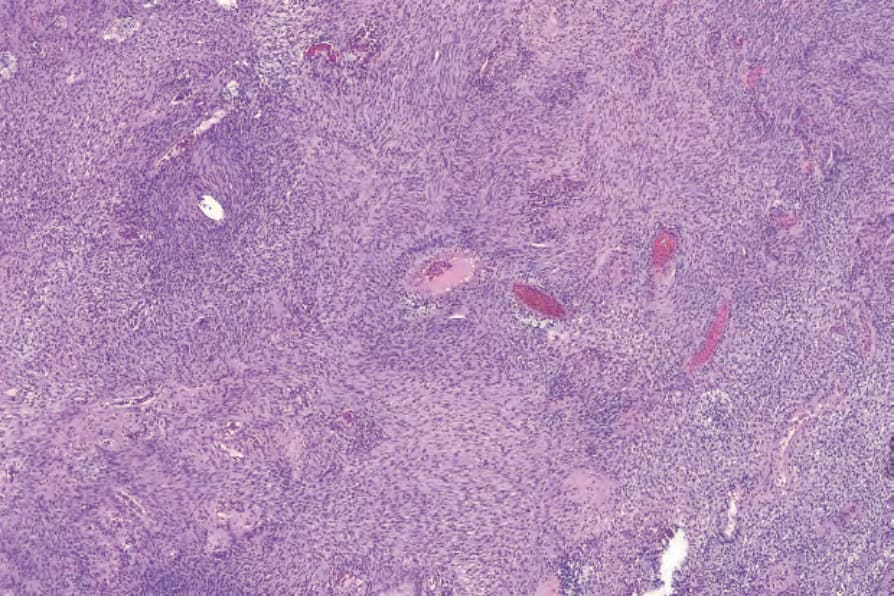
圖 35-317:細胞型神經鞘瘤 (cellular schwannoma):病灶典型上細胞密度高,表面上類似 leiomyosarcoma。
Fig. 35.317 Cellular schwannoma: typically, lesions are highly cellular, superficially resembling leiomyosarcoma.
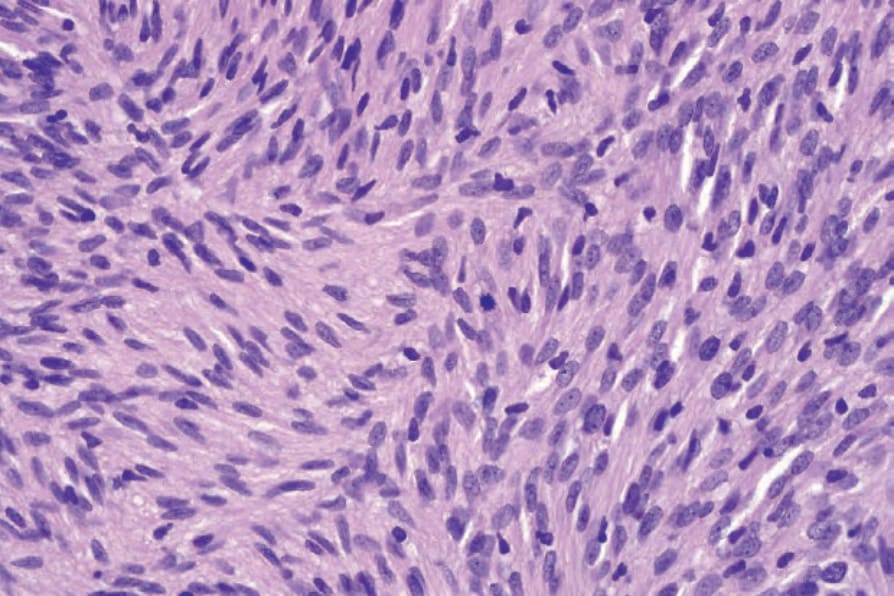
圖 35-318:細胞型神經鞘瘤 (cellular schwannoma):散布的有絲分裂常見。
Fig. 35.318 Cellular schwannoma: scattered mitoses are commonly present.
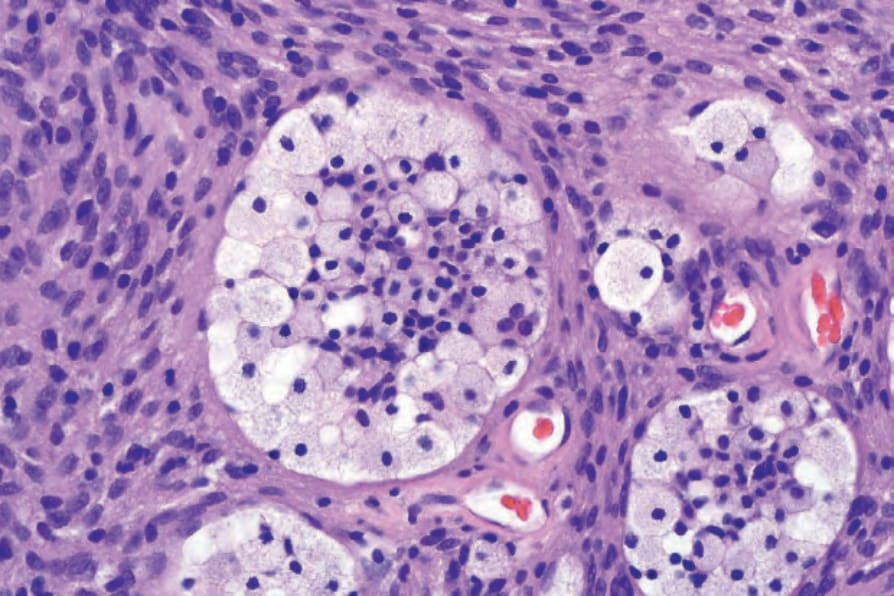
圖 35-319:細胞型神經鞘瘤 (cellular schwannoma):雖然黃色瘤樣組織球 (xanthomatous histiocytes) 可見於任何型別的 schwannoma,但在此變異型中特別常見。
Fig. 35.319 Cellular schwannoma: although xanthomatous histiocytes may be seen in any type of schwannoma, they are particularly common in this variant.
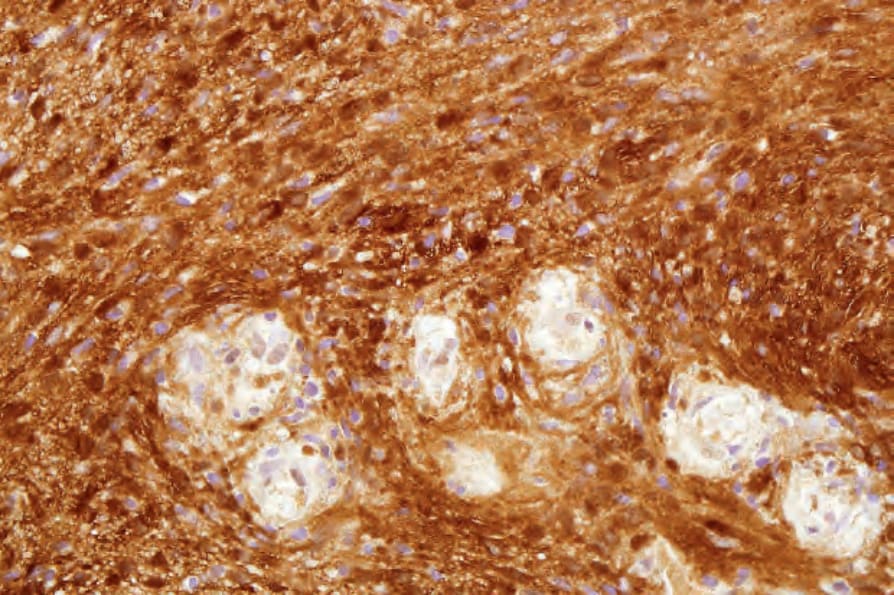
圖 35-320:細胞型神經鞘瘤 (cellular schwannoma):梭形細胞呈 S100 陽性。
Fig. 35.320 Cellular schwannoma: the spindled cells are S100 positive.
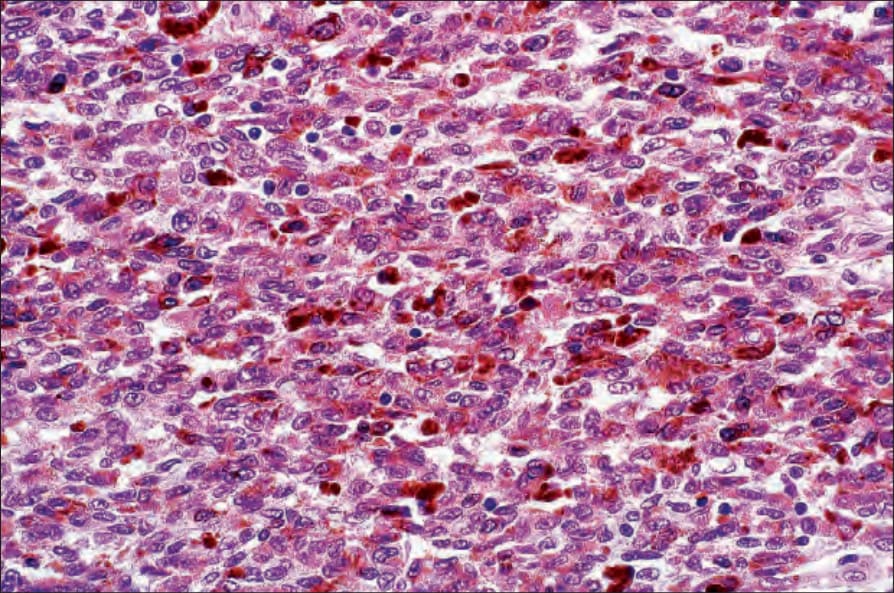
圖 35-321:惡性黑色素性許旺氏腫瘤 (malignant melanotic schwannian tumor):通常為深部病灶,顯示廣泛的黑色素沉著 (melanin pigmentation)。核常有溝紋 (grooved),類似咖啡豆。
Fig. 35.321 Malignant melanotic schwannian tumor: this is usually a deep-seated lesion showing extensive melanin pigmentation. The nuclei are often grooved, resembling coffee beans.
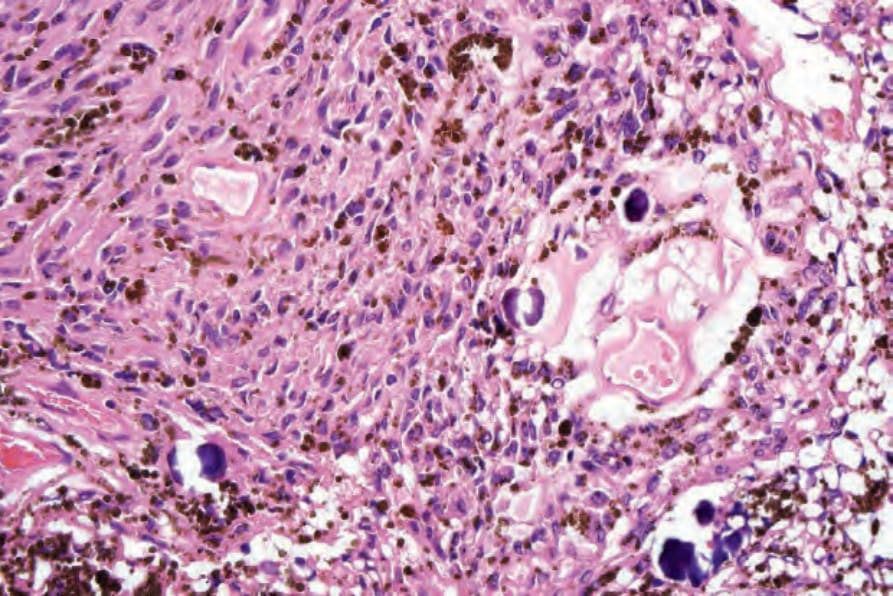
圖 35-322:惡性黑色素性許旺氏腫瘤 (malignant melanotic schwannian tumor):砂粒體 (psammoma bodies) 為有用的線索,但在此腫瘤中並非總是可見。
Fig. 35.322 Malignant melanotic schwannian tumor: psammoma bodies are a useful clue but are not always seen in this tumor.
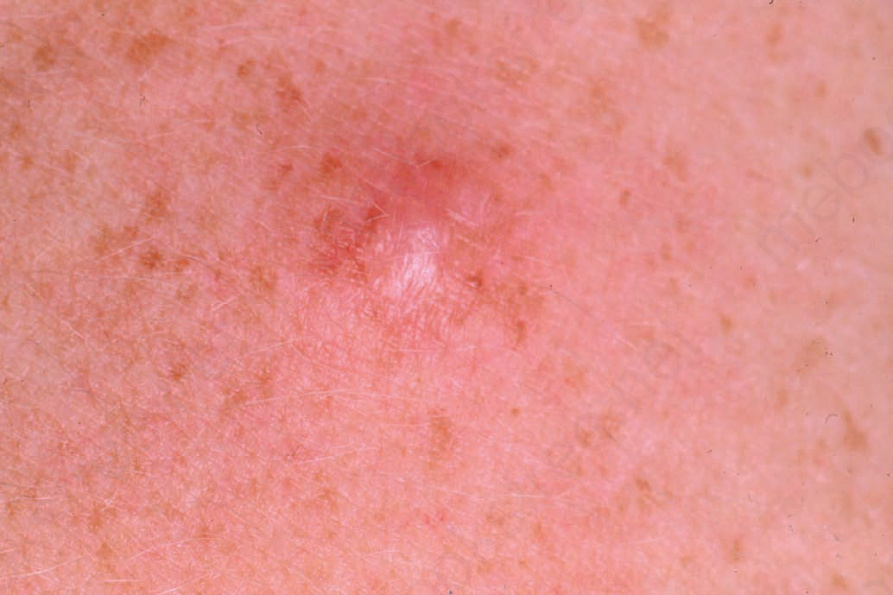
圖 35-323:神經纖維瘤 (neurofibroma):紅斑性結節,周圍環繞單純性雀斑樣痣 (simple lentigines)。取自已故 N.P. Smith, MD, Institute of Dermatology, London, UK 之收藏。
Fig. 35.323 Neurofibroma: erythematous nodule with surrounding simple lentigines. From the collection of the late N.P. Smith, MD, Institute of Dermatology, London, UK.
過去被視為 pacinian neurofibroma,但其組織學特徵更符合 schwannoma。
• 上皮樣神經鞘瘤 (Epithelioid schwannoma) 與神經母細胞瘤樣神經鞘瘤 (neuroblastoma-like schwannoma) 為 schwannoma 的罕見變異型,主要表現於成人的皮下或真皮,無性別偏好,且顯示重疊的特徵。116–120 深部病灶(主要為肌肉內或胃腸道)非常罕見。116,117 與 schwannomatosis 1 相關屬例外。116 大多數病灶為小型,僅少數可觀察到非常大的腫瘤。116 好發於四肢,其次為軀幹。116,117 組織學上,腫瘤界限清楚,由上皮樣細胞 (epithelioid cells) 的局灶巢 (focal nests) 組成,其雙嗜性 (amphophilic) 細胞質排列成索狀與巢狀,伴隨玻璃樣變或黏液樣基質。有時可見具典型 schwannoma 特徵的區域。有絲分裂活性通常較低。標示為非典型 (atypical) 的案例,定義為高有絲分裂活性(等於或大於每 10 HPFs 3 個有絲分裂)與核大小的變異。腫瘤細胞對 S100 與 SOX10 呈陽性,且 collagen type IV 描繪出個別單元或巢中的細胞。116,117 GFAP 為不定陽性。Melan-A 與 keratin 屬例外性陽性。116 包膜含有 EMA 陽性細胞。約 42% 的腫瘤在免疫組織化學上顯示 SMARCB1/INI1 缺失。117 在 neuroblastoma-like schwannoma 中,上皮樣區域可能模擬 neuroblastoma,並含有具纖維性膠原中心 (fibrillary collagenous centers) 的玫瑰花結樣結構 (rosette-like structures)。121 然而,腫瘤的其他區域則為典型的 schwannoma。病灶罕見可呈叢狀。122 已記載一例帶有明顯膠原沉積的案例,以及一例兼具 perineurioma 特徵的混合型腫瘤。123,124 其行為為良性,即使在標示為非典型的案例中,幾乎無局部復發風險。116,117 顯示轉化為惡性上皮樣神經鞘瘤 (malignant epithelioid schwannoma) 的腫瘤非常罕見。117
• 微囊性/網狀神經鞘瘤 (Microcystic/reticular schwannoma) 通常表現於胃腸道,但一部分案例發生於皮膚,呈多分葉性增生,具微囊性、網狀、蕾絲樣 (lace-like) 或假腺樣 (pseudoglandular) 模式,背景有明顯的黏液樣與/或黏蛋白性物質。125,126 腫瘤細胞對 S100 protein 顯示瀰漫性陽性、對 GFAP 顯示不定陽性,且腫瘤分葉周邊可見不連續的 EMA 陽性神經束膜 (perineurium)。
發生率。1,2 病灶最常侵犯下肢,其次為上肢,少數腫瘤發生於頭頸部與軀幹。罕見報告的部位包括胸膜 (pleural) 與內耳道 (internal auditory canal)。3,4 曾有一例報告可能與輻射相關。5 它與 neurofibromatosis 無關。它為良性,少有局部復發傾向。6,7 惡性轉化屬例外。單純切除為首選治療。
組織學特徵 (Histologic Features)
腫瘤界限清楚,但缺乏包膜,由溫和的梭形細胞 (bland spindled cells) 組成,其細胞質淡染、界限不清,核呈拉長、末端漸尖。腫瘤細胞的分布模式為輪輻狀 (storiform)、板層狀 (lamellar) 或漩渦狀 (whorled)。可出現黏液樣變化,亦可出現似為退化性質的局部細胞學異型 (cytologic atypia)。Antoni A 與 Antoni B 區並非其特徵。有絲分裂相罕見。浸潤性生長模式與叢狀結構屬例外;已有報告帶有上皮樣神經鞘瘤 (epithelioid schwannoma) 成分的案例。8,9 一例腫瘤被鑑定發生於先天性黑色素細胞痣 (congenital melanocytic nevus) 中。10 藉由雙重染色 (double staining),已證實腫瘤細胞不是 Schwann cells 就是神經束膜細胞 (perineural cells),以前者為主,且無抗原共表現 (no antigen coexpression)。1,2 Schwann cells 對 S100 protein 與 SOX10 呈陽性,而神經束膜細胞表現 EMA 與 CD34;腫瘤細胞通常對 CD34、GFAP 與 claudin 1 呈陽性。軸突 (Axons) 罕由 neurofilament 標示。1,2,7,11–13