Schwannoma
Schwannoma
Clinical features Schwannomas (neurilemmomas) are common benign lesions, occurring most often in the fourth and fifth decades with an equal sex incidence and arising most frequently on the limbs (mainly the upper limbs) followed by the head and neck (including the oral cavity, orbit and salivary glands) (Fig. 35.308).1–5
1785 Benign neural tumors
Lesions in children are very rare and exceptionally congenital.6,7 They present most often as a solitary painless subcutaneous mass of variable size (exceptionally very large), but very occasionally they may be multiple and in this context are rarely associated with von Recklinghausen neurofibromatosis.8,9 Prominent cystic change is occasionally seen. Purely dermal tumors are rare.10,11 Cutaneous lesions exceptionally have an agminate pattern and one was associated with overlying anetoderma.12,13 Tumors in the penis and vulva are exceedingly rare.14,15 Some tumors occur in other locations including bone, gastrointestinal tract, pancreas, liver, retroperitoneum, mediastinum, trachea, nasopharynx, larynx, thyroid, adrenal gland and lymph node.16–26 Neurological symptoms including pain and paresthesias are uncommon except in large deep-seated lesions; malignant change is exceedingly rare (see neurofibroma).5,27,28 Exceptional cases include a cutaneous example that may have been associated with foreign material.29 Recurrence after simple excision is very infrequent.30
Neurofibromatosis type II (NF2 gene at 22q12.2 encodin merlin protein) is characterized by acoustic schwannomas, cutaneous tumors and other central nervous system lesions including meningioma, cataract and retinal hamartoma.31,32 About 59% of patients have skin tumors, the majority of which represent schwannomas.32 Only rarely do patients develop neurofibromas or hybrid lesions. Café-au-lait spots may be present in up to 33% of patients but these tend to be fewer than in patients with neurofibromatosis type I.32 The development of neurofibromas in neurofibromatosis type II may be due to interaction between neurofibromin and merlin, the NF2 gene product, in regulating the RAS proto-oncogene.33,34 Cutaneous schwannomas are only rarely associated with neurofibromatosis type II.35
The National Institute of Health (NIH) diagnostic criteria for NFII are as follows:
• Bilateral vestibular schwannomas that do not require any additional features for diagnosis of the disease,
• First-degree relative with NF2 and either occurrence of unilateral vestibular schwannoma in patients younger than 30 or occurrence of two other associated lesions (e.g., glioma, meningioma, schwannoma, juvenile cortical cataract),
• Unilateral vestibular schwannoma and any two other associated lesions including glioma, meningioma, schwannoma, neurofibroma, or juvenile cortical cataract,
• Multiple meningiomas with any of the above lesions.23
The presence of multiple cutaneous schwannomas with or without similar lesions in spinal and other nerves has been termed schwannomatosis. Although it was initially doubted whether it represents a discreet entity or merely a variant of neurofibromatosis type II, it is now regarded as distinct.32,36–45 Most cases appear to be sporadic but in a few an autosomal dominant pattern of inheritance has been described. Cases associated with multiple meningiomas and a family with predisposition to malignant rhabdoid tumors have been described.46–50 Germline aberrations in INI1 / SMARCB1 (22q11) may be involved.
A new hereditary syndrome consisting of multiple schwannomas, multiple nevi and multiple vaginal leiomyomas has been described.51 The nevi are congenital but the schwannomas and vaginal leiomyomas develop in adult life.
In a recent study it was found that children or young adults that develop solitary schwannoma or meningioma usually have genetic predisposition.52
Pathogenesis and histologic features Cytogenetic studies in schwannomas have shown either loss of 22q material or monosomy 22, probably corresponding to the NF2 gene (22q12.2) encoding the neurofibromin 2 or merlin protein.34,35,53,54
Schwannomas are usually rounded and invariably encapsulated, and are typically found in the subcutaneous or deeper tissues; primary intradermal origin is unusual. Purely intraneural tumors are exceptional.55 Microscopically, they are characterized by a classical biphasic pattern of cellular Antoni A and hypocellular Antoni B areas.
• Antoni A areas form the more cellular component of the lesion and are composed of fairly closely packed spindled cells with tapering, elongated, rather wavy nuclei; nuclear palisading is a prominent feature, producing the distinctive Verocay bodies (Figs 35.309–35.311). These
1786 Connective tissue tumors
are sometimes the predominant feature.56 Verocay-like bodies may be seen in a number of other tumors including dermatofibroma and leiomyoma.57 Degenerative nuclear pleomorphism and mitotic activity are occasionally seen, but tend to be spatially unrelated. Hyalinization of stromal collagen and focal dystrophic calcification are sometimes present.
• Antoni B areas are typified by irregularly scattered spindled or stellate cells set in an abundant loose myxoid stroma (Fig. 35.312). Within these areas, scattered chronic inflammatory cells and small blood vessels, often with hyalinized walls, are a prominent feature (Fig. 35.313). Focal degenerative changes, including microcystic change and hemosiderin deposition, are not uncommon. A schwannoma with collagenous spherulosis has been documented and in one case meningothelial-like whorls were present.58,59 An intravascular presentation is exceptional.60
The very rare finding of apparent glandular differentiation in benign schwannomas represents proliferation of entrapped normal adnexal structures.61–64
Though relatively rare, tumors exhibiting a hybrid appearance of schwannoma and neurofibroma are increasingly recognized.65–72 These can be seen in patients with neurofibromatosis and often the neurofibroma is of
1787 Benign neural tumors
the plexiform variant.73 They appear to be overrepresented amongst patients with schwannomatosis and neurofibromatosis.74 One hybrid case showed monosomy 22 (loss of NF2 locus) as well as loss of function mutations in the CTNNA3 gene (10q21.3).
The schwannomas seen in neurofibromatosis type II have been shown to contain axons.76
Malignant transformation often shows pleomorphic epithelioid cells and rarely there is divergent differentiation such as the presence of epithelioid angiosarcoma.77–79
Ultrastructurally, schwannomas are composed predominantly of Schwann cells and this is reflected immunohistochemically by S100 protein positivity in the majority of tumor cells. The capsule contains a layer of EMA-positive perineurial fibroblasts. GFAP and CKAE1/AE3 positivity may be seen mainly in deep seated tumors. SOX10, podoplanin and calretinin are positive in the majority of cases whereas positivity for CD34 is more focal.80–85 Tumor cells express PDGFR-alpha, PDGFR-beta ligands and their cognate receptors as well as KIT, and it has been shown that imatinib mesylate inhibits a schwannoma cell line.86,87
Variants88
• Plexiform schwannoma is an uncommon tumor only very rarely associated with neurofibromatosis (mainly NF2) and tending to arise mainly on the head and neck or trunk of children or young adults.89–98 These lesions represent about 4.3% of all schwannomas and around 15% of cutaneous schwannoma. It is usually a small intradermal or subcutaneous lesion characterized by multiple encapsulated nodules composed predominantly of Antoni A tissue (Fig. 35.314). Any histologic type of schwannoma may be represented in plexiform tumors, particularly the cellular variant. Cellular plexiform schwannoma occurs in infants and recurrence of trisomy 17 has been reported.98 It shows lack of circumscription or an infiltrative growth pattern, increased cellularity and relatively high mitotic activity. These features may lead to a diagnosis of malignancy especially in small biopsies. There is no metastatic potential but local recurrence is not unusual.89,98
• A subgroup of plexiform and multinodular schwannomas affects major peripheral nerves. Deep-seated tumors also occur. Nuclear pleomorphism (mild to moderate), limited mitotic activity and focal necrosis (the latter in deep-seated examples) may be present, but recurrence is not a feature and there is no malignant potential. Distinction from plexiform neurofibroma is vital to avoid an inappropriate clinical diagnosis of von Recklinghausen disease (neurofibromatosis type 1). Rarely, tumors are associated with neurofibromatosis type 2 and schwannomatosis.
• Ancient schwannoma, which is usually a more deeply located, long-standing lesion, is characterized by pronounced degenerative changes manifest as nuclear pleomorphism associated with extensive cyst formation, calcification, hyalinization or hemorrhage (Figs 35.315 and 35.316).99 Mitoses, however, are rare.
• Cellular schwannoma only rarely presents subcutaneously as a large and encapsulated mass.100-–102 Microscopically, there is a marked increase in cellularity which, combined with a mainly fascicular architecture, may simulate a smooth muscle tumor (Figs 35.317 and 35.318). Verocay bodies are generally not seen. Xanthomatous cells and a lymphocytic infiltrate may be prominent (Fig. 35.319). Normal mitoses may number up to 10 per 10 high-power fields, but neither necrosis nor significant nuclear pleomorphism is a feature. Distinction from smooth muscle tumors is readily afforded by S100 positivity (Fig. 35.320).
• Malignant melanotic schwannian tumor (previously known as melanotic schwannoma, psammomatous melanotic schwannoma) is a rare lesion which, in addition to the features of a neural tumor, contains pigmented cells and usually displays psammoma bodies (Fig. 35.321).103,104 Based on the observation that tumors recur and may metastasize, it has recently been proposed that all lesions should be
1788 Connective tissue tumors
labelled as malignant melanotic schwannian tumor. It arises most frequently around the spinal nerve roots and cutaneous presentation is very rare, with only 20 cases reported so far arising in the dermis or subcutaneous tissue, two of which have developed metastasis, and one patient died of disseminated disease.105,106 Two cases arising in association with nevus of Ota have been documented.107 The tumor is usually seen in association with Carney complex (myxomas, spotty pigmentation, and endocrine overactivity; autosomal dominant with mutation of PRKAR1A) (Fig. 35.322).108,109 Lesions associated with the latter may also present with metastatic disease.110 Tumor cells stain for S100 protein, SOX10 and other melanocytic markers. CD34 may also be positive.111 Exceptionally a tumor may display a plexiform pattern.112 Histologic features do not allow prediction of behavior, except for a mitotic rate of greater than 2 per 10 HPF, which correlates with metastases.103,113
• Pacinian schwannoma is a very rare tumor that presents as a solitary nodule, most often in the distal extremities. It is characterized histologically by an encapsulated mass composed of round or ovoid concentrically lamellated corpuscles (somewhat resembling pacinian corpuscles) set in a collagenous spindled cell stroma.114,115 Although
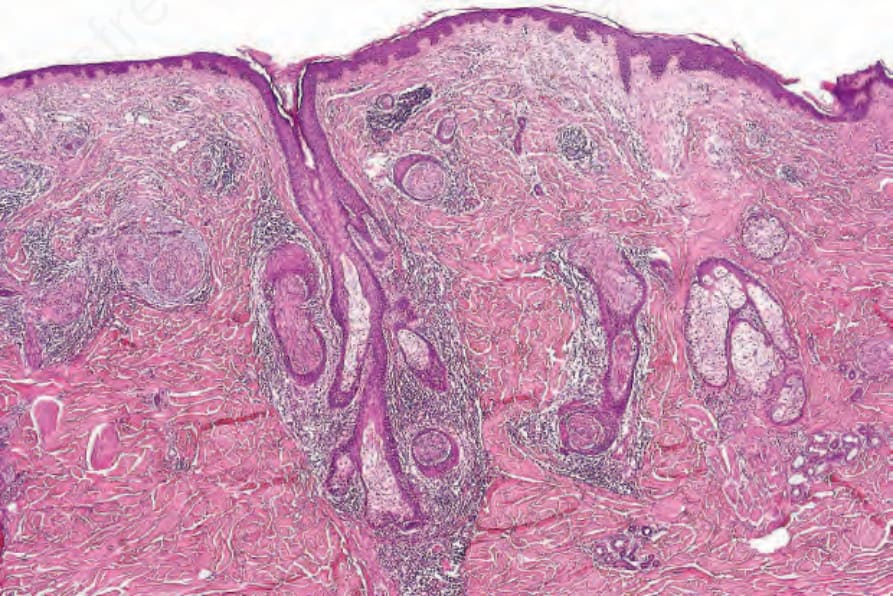
Fig. 35.306 Epithelial sheath neuroma: prominent nerves are present in the superficial reticular dermis encased by nests of bland squamous epithelium. By courtesy of L. Requena, MD, Madrid, Spain.
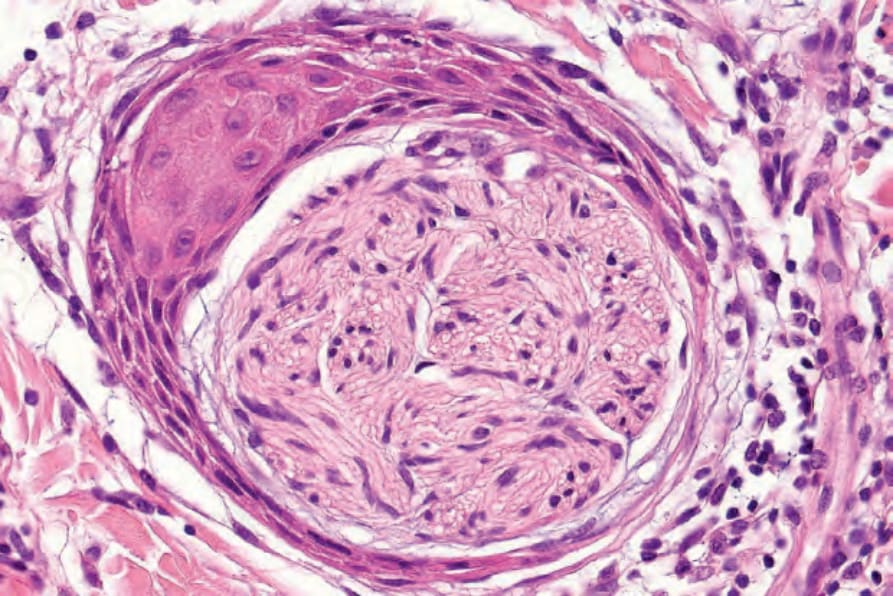
Fig. 35.307 Epithelial sheath neuroma: high-power view. By courtesy of L. Requena, MD, Madrid, Spain.
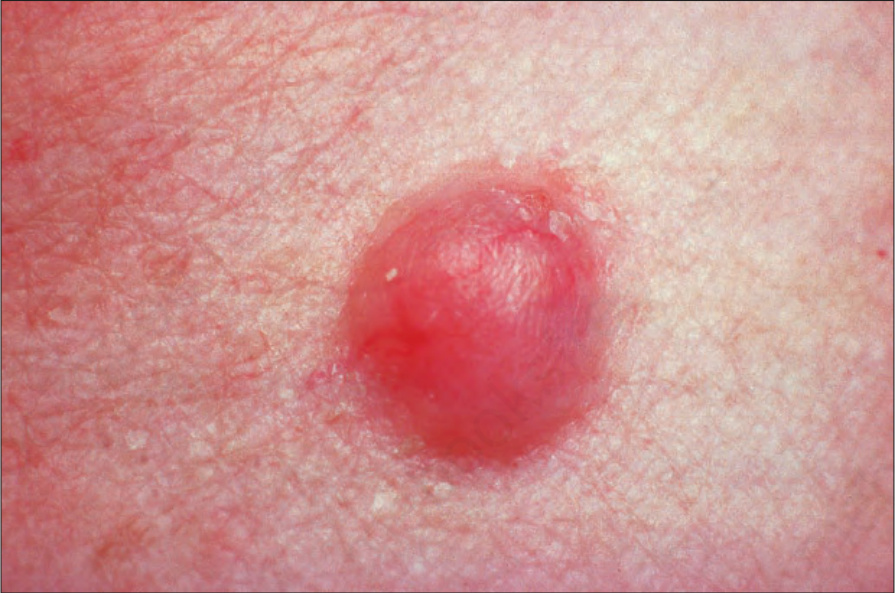
Fig. 35.308 Schwannoma: this tumor presents as a non-specific dermal nodule. By courtesy of the Institute of Dermatology, London, UK.
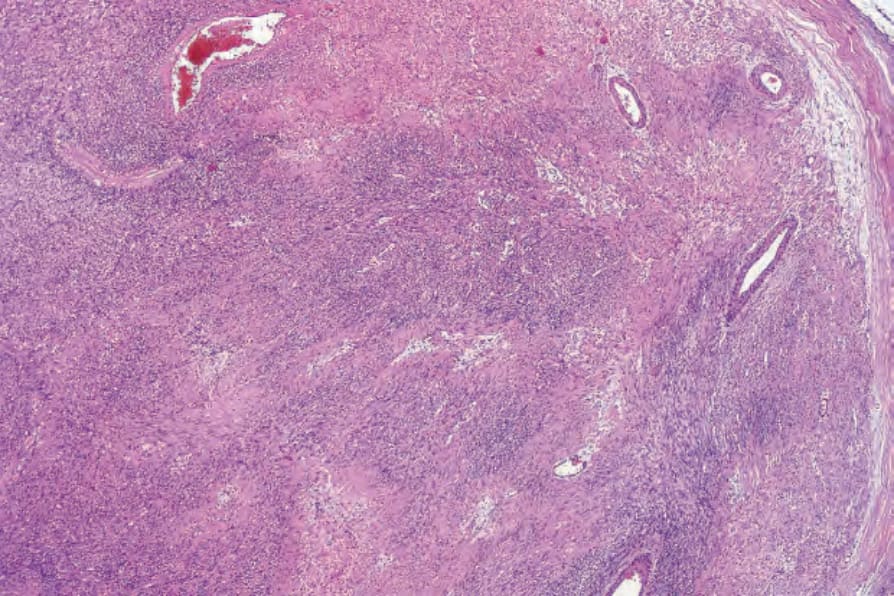
Fig. 35.309 Schwannoma: scanning view of spindle cell tumor with prominent blood vessels. A capsule is seen on the right side.
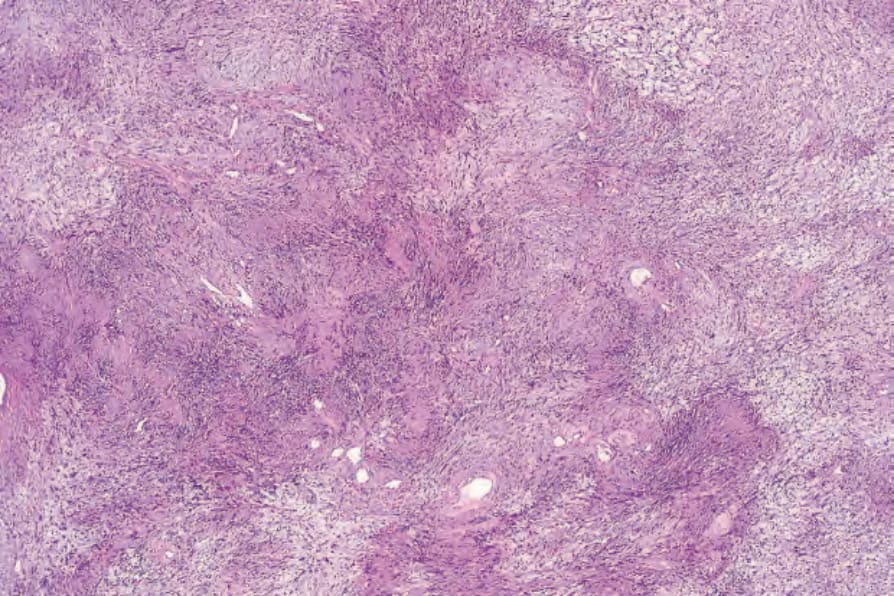
Fig. 35.310 Schwannoma: palisading is a characteristic feature.
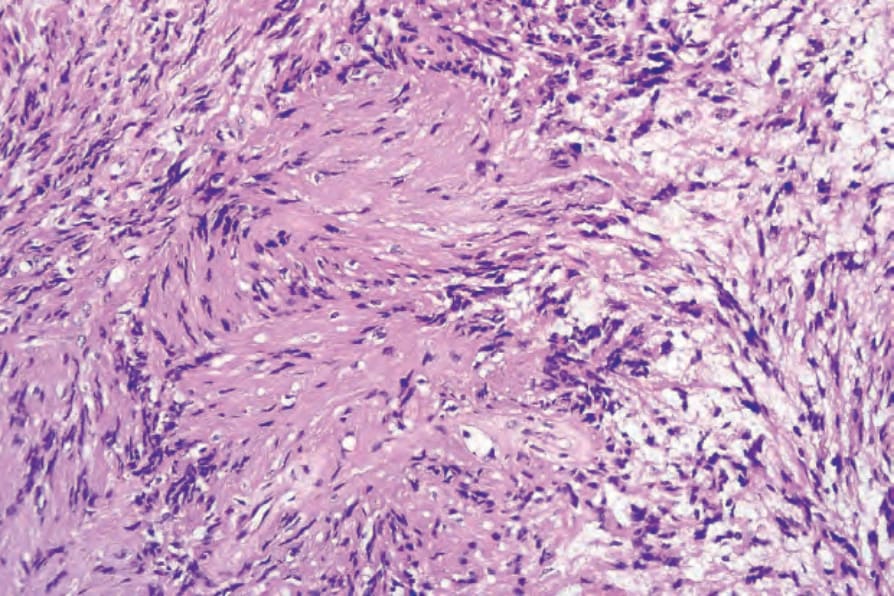
Fig. 35.311 Schwannoma: the Verocay body, typical of the Antoni A areas, is characterized by two parallel rows of nuclei separated by Schwann cell processes.
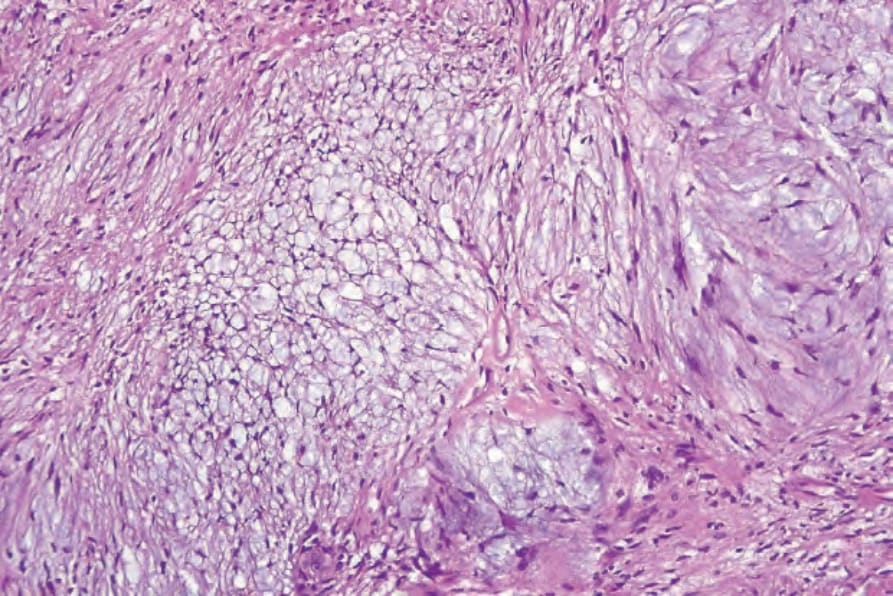
Fig. 35.312 Schwannoma: myxoid degeneration gives rise to Antoni B areas.
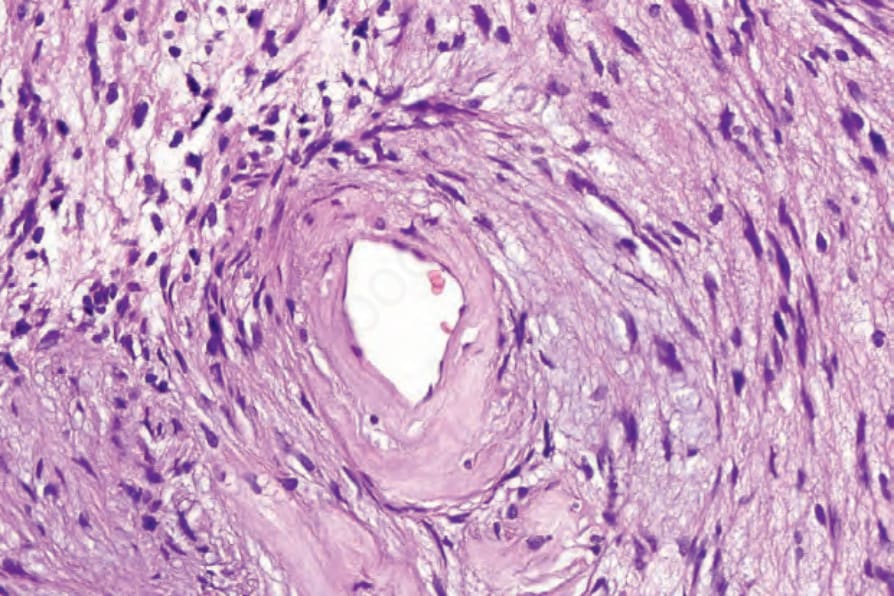
Fig. 35.313 Schwannoma: there is marked hyalinization of the blood vessel walls in Antoni B.
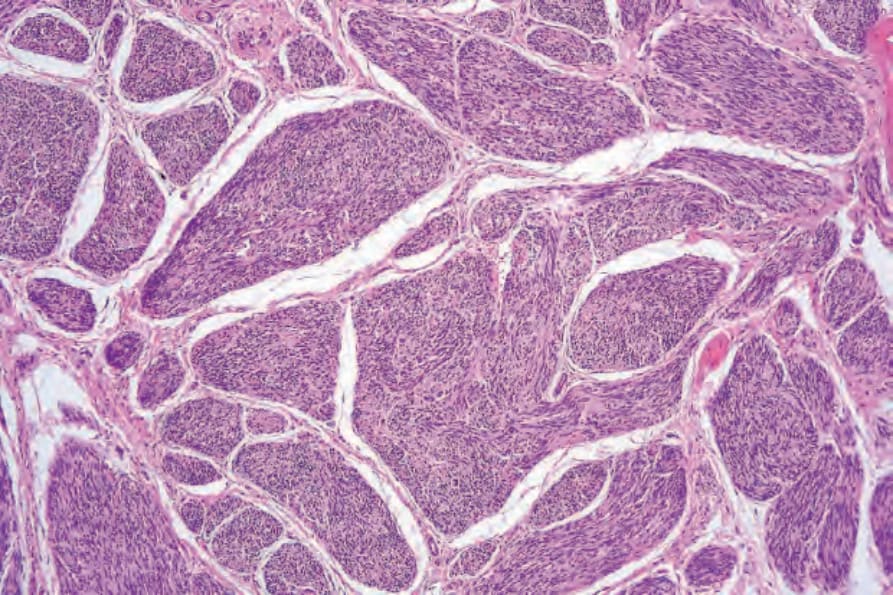
Fig. 35.314 Plexiform schwannoma: this small tumor is composed of multiple discrete nodules of schwannomatous tissue. Nuclear palisading and Verocay bodies are evident.
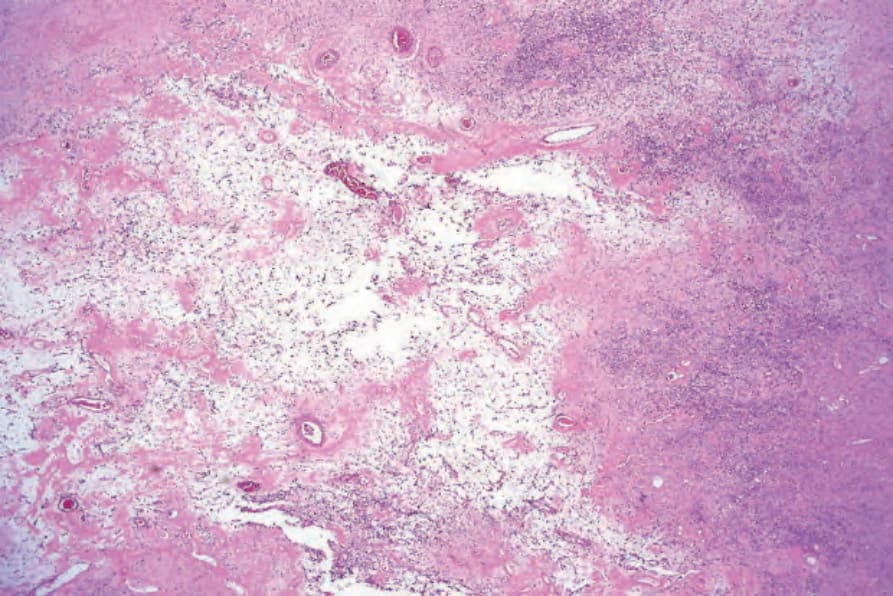
Fig. 35.315 Ancient schwannoma: degenerative changes have resulted in marked myxoid features with fibrosis and conspicuous vascularity.
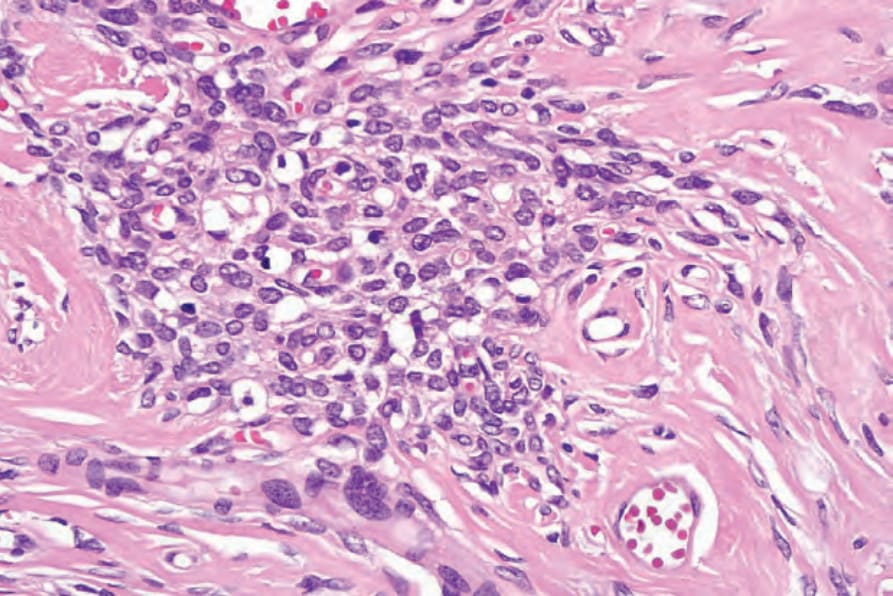
Fig. 35.316 Ancient schwannoma: focal nuclear pleomorphism should not be taken as having sinister implication. Mitotic activity is not present in these tumors.
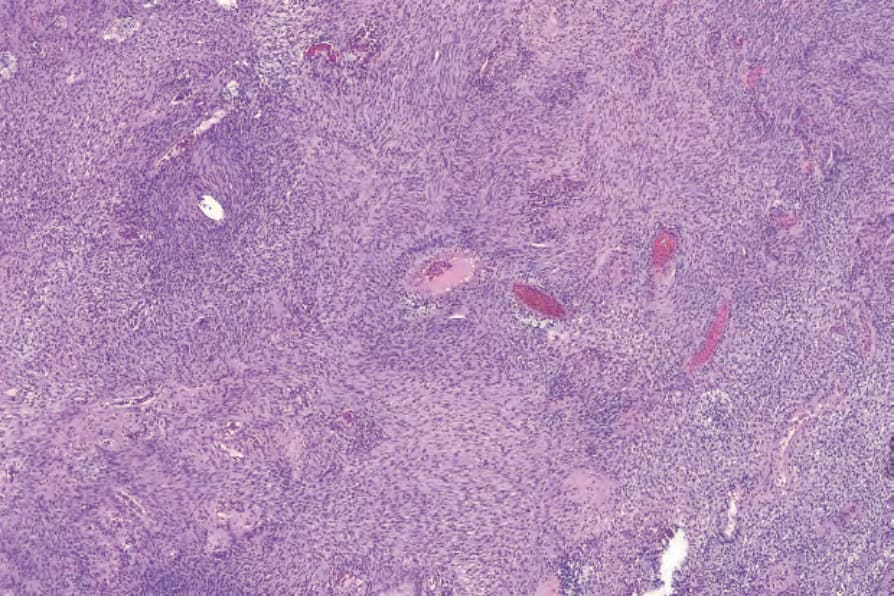
Fig. 35.317 Cellular schwannoma: typically, lesions are highly cellular, superficially resembling leiomyosarcoma.
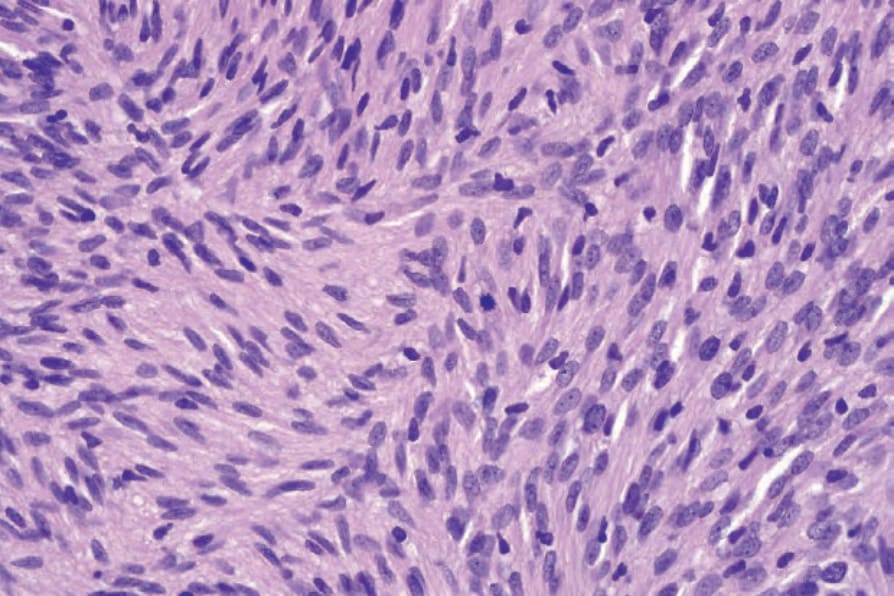
Fig. 35.318 Cellular schwannoma: scattered mitoses are commonly present.
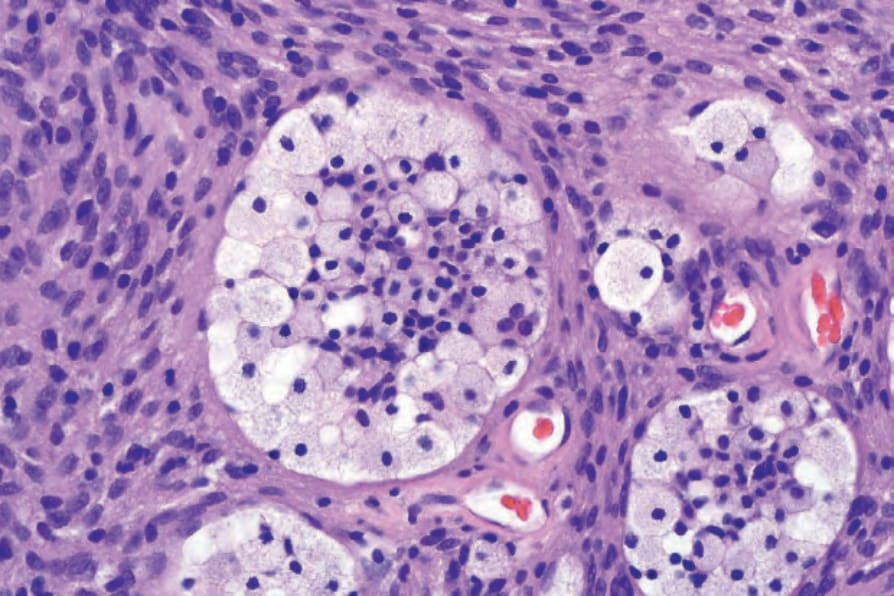
Fig. 35.319 Cellular schwannoma: although xanthomatous histiocytes may be seen in any type of schwannoma, they are particularly common in this variant.
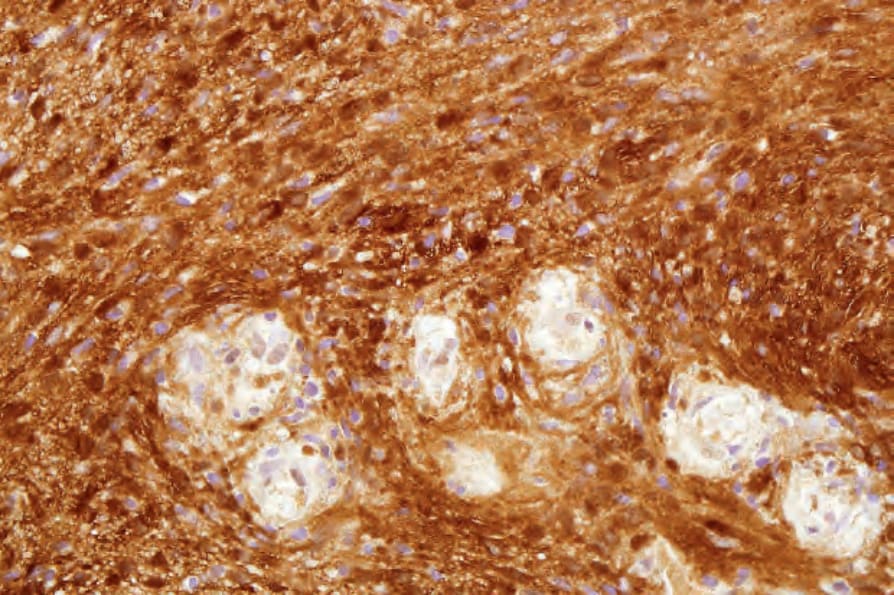
Fig. 35.320 Cellular schwannoma: the spindled cells are S100 positive.
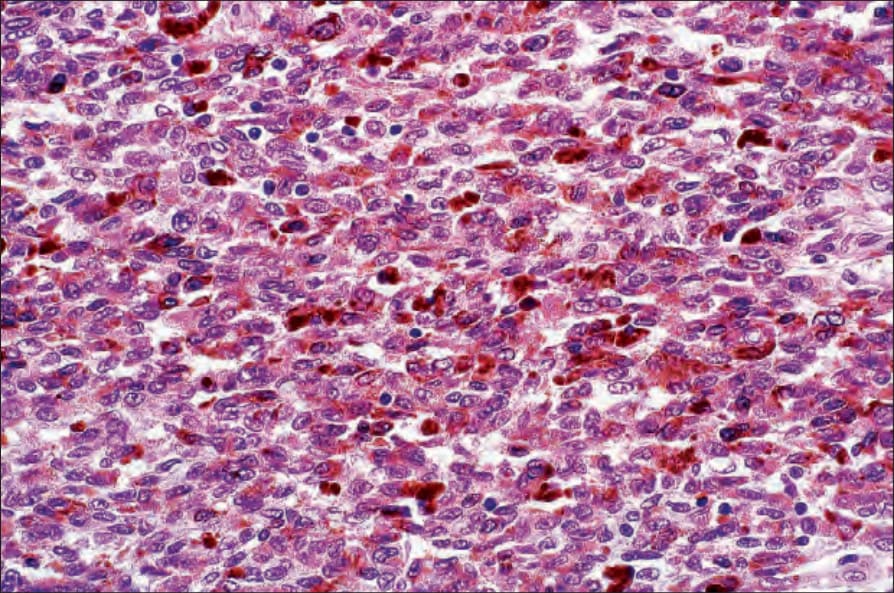
Fig. 35.321 Malignant melanotic schwannian tumor: this is usually a deep-seated lesion showing extensive melanin pigmentation. The nuclei are often grooved, resembling coffee beans.
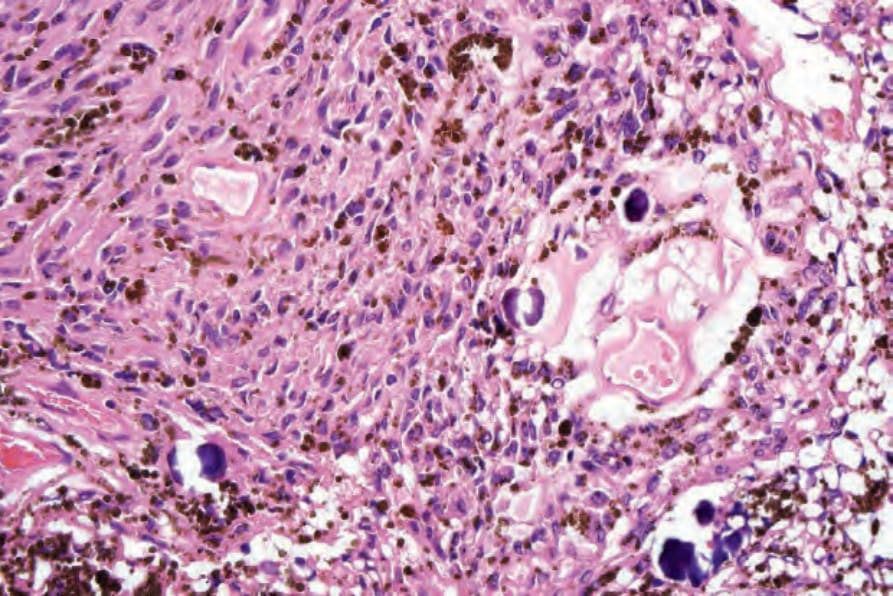
Fig. 35.322 Malignant melanotic schwannian tumor: psammoma bodies are a useful clue but are not always seen in this tumor.
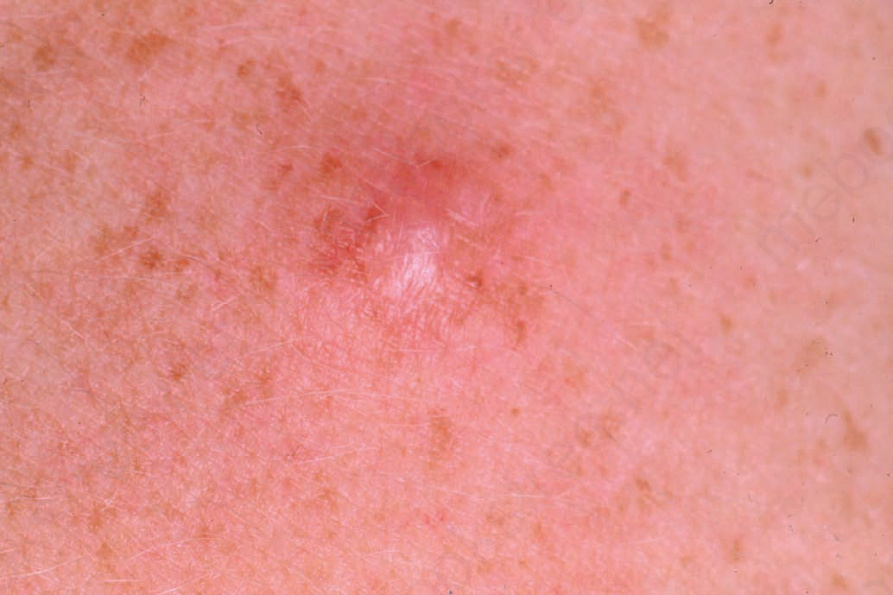
Fig. 35.323 Neurofibroma: erythematous nodule with surrounding simple lentigines. From the collection of the late N.P. Smith, MD, Institute of Dermatology, London, UK.
1789 Benign neural tumors
regarded in the past as pacinian neurofibroma, the histologic features are more in keeping with a schwannoma.
• Epithelioid schwannoma and neuroblastoma-like schwannoma are rare variants of schwannoma presenting mainly in the subcutis or dermis of adults with no sex predilection, and show overlapping features.116–120 Deep-seated lesions, mainly intramuscular or gastrointestinal, are very rare.116,117 An association with schwannomatosis 1 is exceptional.116 Most lesions are small and only rarely very large tumors are observed.116 There is predilection for the limbs followed by the trunk.116,117 Histologically, tumors are well-circumscribed and composed of focal nests of epithelioid cells with amphophilic cytoplasmic arranged in cords and nests associated with a hyalinized or myxoid stroma. Areas with features of classic schwannoma are sometimes seen. Mitotic activity is usually low. Cases labelled as atypical are defined by high mitotic activity (equal or more than 3 mitoses per 10 HPFs) and variation in nuclear size. Tumor cells are positive for S100 And SOX10 and collagen type IV decorates cells in individual units or nests.116,117 GFAP is variably positive. Melan-A and keratin are exceptionally positive.116 The capsule contains EMA-positive cells. Around 42% of tumors show loss of SMARCB1/INI1 by immunohistochemistry.117 In neuroblastoma-like schwannoma the epithelioid areas may mimic neuroblastoma and contain rosette-like structures with fibrillary collagenous centers.121 Other areas of the tumor, however, are typical of a schwannoma. Lesions may rarely be plexiform.122 A case with prominent collagen deposition and a hybrid tumor with perineurioma features have been documented.123,124 The behavior is benign with hardly any risk of local recurrence even in cases labelled as atypical.116,117 Tumors displaying transition to malignant epithelioid schwannoma are very rare.117
• Microcystic/reticular schwannoma usually presents in the gastrointestinal tract but a subset of cases occur in the skin as a multilobular proliferation with a microcystic, reticular, lace-like, or pseudoglandular pattern with prominent myxoid and/or mucinous material in the background.125,126 Tumor cells show diffuse positivity for S100 protein, variable positivity for GFAP, and a discontinuous EMA-positive perineurium may be seen at the periphery of tumor lobules.
incidence.1,2 Lesions most commonly involve the lower limb followed by the upper limbs, with rare tumors occurring in the head and neck and trunk. Rare reported sites include pleural and internal auditory canal.3,4 A possible association with radiation has been reported in one case.5 It is not associated with neurofibromatosis. It is benign with little tendency for local recurrence.6,7 Malignant transformation is exceptional. Simple excision is the treatment of choice.
Histologic features Tumors are well circumscribed but lack a capsule and are composed of bland spindled cells with ill-defined pale cytoplasm and elongated nuclei with tapering ends. The distribution pattern of tumor cells is storiform, lamellar or whorled. Myxoid change can be present as can focal cytologic atypia that appears to be degenerative in nature. Antoni A and Antoni B areas are not a feature. Mitotic figures are rare. An infiltrative growth pattern and plexiform architecture are exceptional; cases with an epithelioid schwannoma component have been reported.8,9 In one case a tumor was identified in a congenital melanocytic nevus.10 By double staining, it has been demonstrated that tumor cells are either Schwann cells or perineural cells, with predominance of the former and no antigen coexpression.1,2 Schwann cells are positive for S100 protein and SOX10 whereas perineural cells express EMA and CD34; tumor cells are usually positive for CD34, GFAP and claudin 1. Axons are rarely highlighted by neurofilament.1,2,7,11–13