疾病定義與臨床特徵
- 血管免疫母細胞型 T 細胞淋巴瘤 (Angioimmunoblastic T-cell lymphoma, AITL) 為一種周邊 T 細胞淋巴瘤 (peripheral T-cell lymphoma),表現為全身性疾病,包括廣泛淋巴結病變 (generalized lymphadenopathy)、肝脾腫大 (hepatosplenomegaly)、貧血與高免疫球蛋白血症 (hypergammaglobulinemia)。
- 男女比變異不定,發病年齡多在第六或第七個十年。
- 通常於就診時已屬晚期,伴全身性淋巴結病變、常見肝脾腫大、骨髓與皮膚侵犯及全身症狀;亦可見肋膜積液、腹水、關節炎/關節痛、耳鼻喉侵犯與神經學表現。
- 常見溶血性 (Coombs 陽性) 貧血、多株性高免疫球蛋白血症、LDH 上升、白血球增多 (含嗜酸性球增多) 或淋巴球減少與血小板減少。
- 可伴多種自體免疫現象:冷凝集素、冷球蛋白、循環免疫複合體、平滑肌抗體、類風濕因子、抗核抗體。
- 皮膚病灶見於高達 50% 病人 (就診時及/或復發時),表現為軀幹與四肢的搔癢性斑丘疹 (maculopapular eruption),可類似病毒疹或藥物過敏反應,部分病灶於用藥後出現;其他表現包括蕁麻疹、紅皮症、糜爛、瘀點、紫斑、丘疹水疱性癢疹樣病灶、斑塊與腫瘤結節。
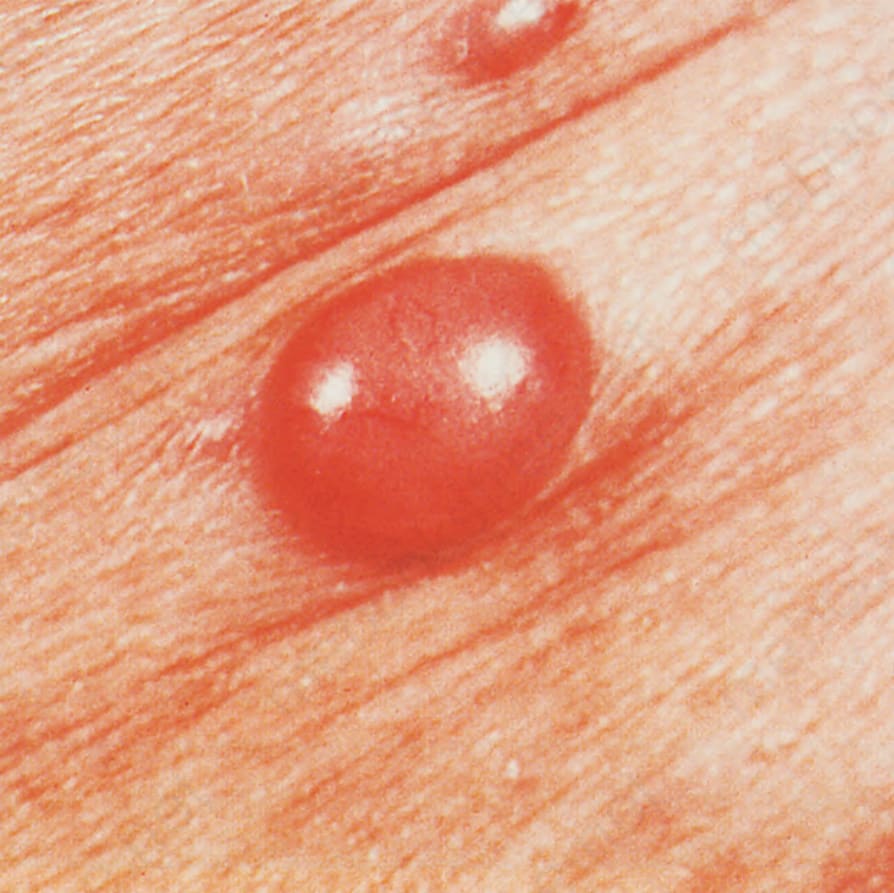
圖 29-141:血管免疫母細胞型 T 細胞淋巴瘤,前胸壁與前腋窩皺褶處的紅斑性斑丘疹。
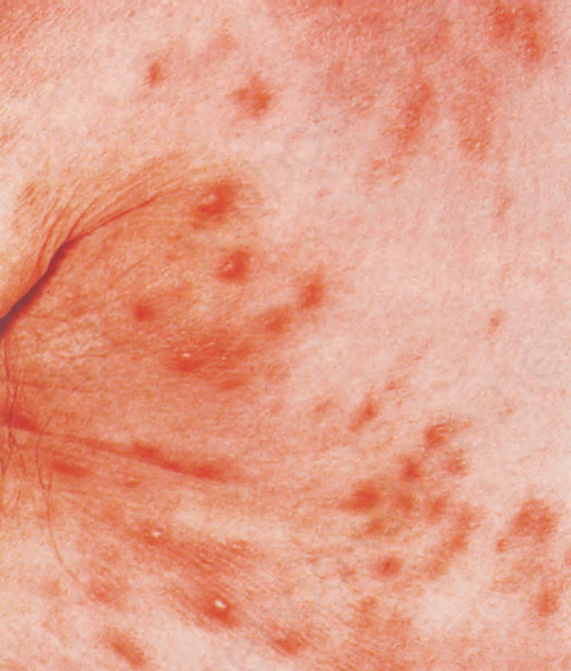
圖 29-142:血管免疫母細胞型 T 細胞淋巴瘤,亦出現結節性沉積。
致病機轉與細胞遺傳學
- AITL 為濾泡輔助 T 細胞 (follicular helper T cells) 的惡性腫瘤,由免疫表型與基因表現譜佐證。
- 最常見染色體異常為第 3、5、18、19 號染色體三倍體、X 染色體增益與第 7 號染色體缺失;CGH 另顯示部分病例有 11q13、19、22p 增益及 13q 缺失。
- 表觀遺傳調控基因突變常見:RHOA、TET2、DNMT3、IDH2;其中僅 IDH2 突變對此亞型相對具特異性。
組織病理特徵
- 淋巴結:典型呈結構部分消失,由多形性浸潤 (polymorphic infiltrate) 取代,主要位於副皮質區 (paracortical areas);竇通常保留,結周結締組織常受侵犯。早期可見增生性 B 細胞濾泡,但濾泡常退縮或消失。副皮質區可見高內皮小靜脈 (high endothelial venules) 明顯增生,及淋巴球、漿細胞、嗜酸性球與組織球混合族群;腫瘤淋巴球多為中等大小,胞質透明或淡染,常聚集於濾泡與高內皮小靜脈周圍。亦見散在大型 B 免疫母細胞,部分類似 Reed-Sternberg 細胞。
- 皮膚切片:多見淺層或淺深層血管周圍 (偶見腺體周圍) 淋巴球浸潤,含多形性非典型淋巴球並伴血管增生與內皮細胞突顯;較少見者為非特異性的淺層血管周圍小淋巴球浸潤、嗜酸性球與漿細胞。部分病例可見白血球破碎性血管炎與肉芽腫性發炎。罕見情況下伴隨的 (有時為腫瘤性) B 細胞擴張可主導組織像,掩蓋潛在的 T 細胞淋巴瘤;此 B 細胞增生多類似瀰漫性大 B 細胞淋巴瘤 (DLBCL)。
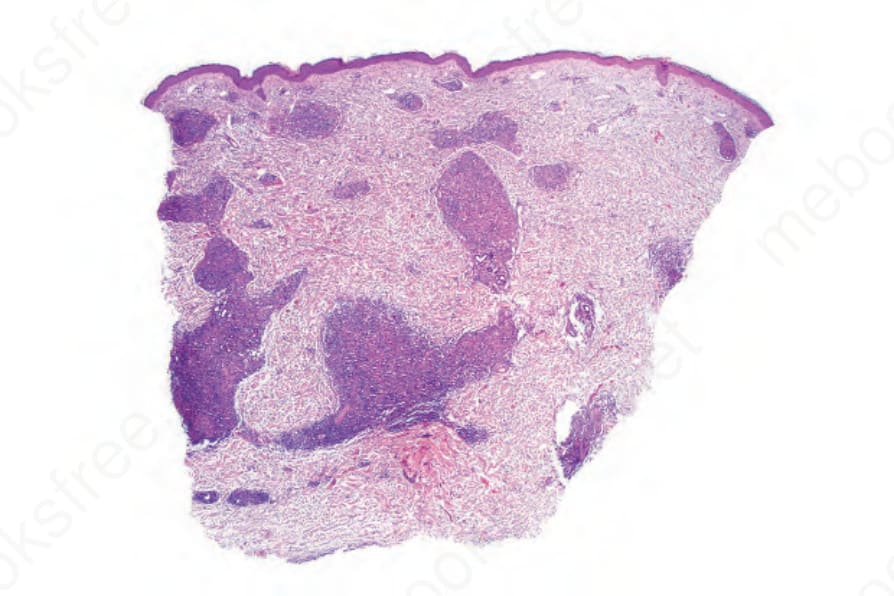
圖 29-143:血管免疫母細胞型 T 細胞淋巴瘤,切片顯示明顯的肉芽腫性真皮浸潤。
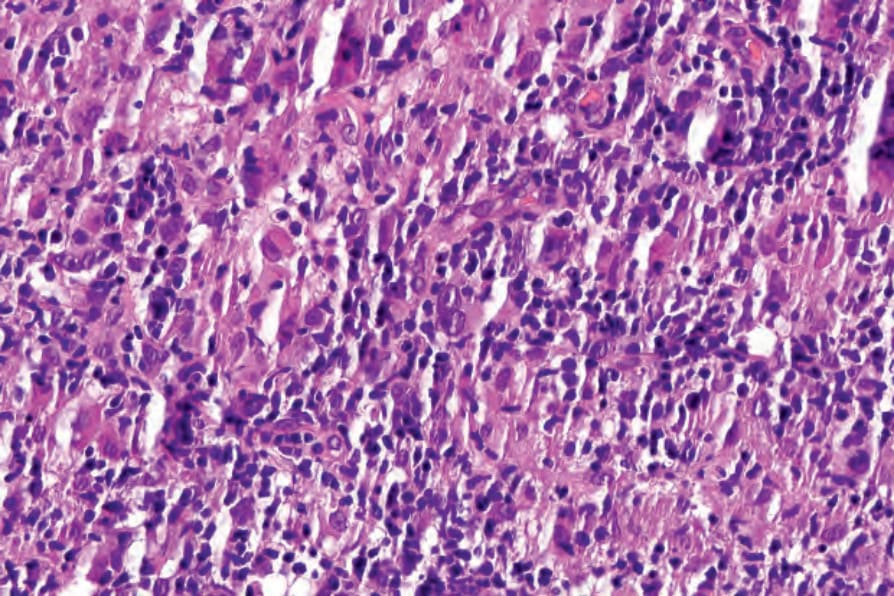
圖 29-144:血管免疫母細胞型 T 細胞淋巴瘤,可見眾多組織球與散在非典型細胞。
免疫表型與分子檢查
- 淋巴結內腫瘤細胞為 CD4 陽性,通常表現 CD2、CD3、CD5;部分表現濾泡輔助細胞標記 CD10、CXCL13、PD1,有時 bcl6。CD20 標示 B 免疫母細胞,後者常 EBV 原位雜交陽性。CD21、CD23、CD35 及/或 CNA42 標示不規則擴大的濾泡樹突細胞網。
- 皮膚中腫瘤淋巴球辨識較難,CXCL13 抗體免疫組化具敏感性與特異性。
- 多數病例淋巴結可測得 TCR 基因克隆性重組,且與皮膚 CXCL13 陽性腫瘤細胞相關。20–30% 病例見克隆性免疫球蛋白基因重組,對應 EBV 陽性 B 細胞的擴增族群。
鑑別診斷
- 區分皮膚 AITL 侵犯與反應性皮膚浸潤,最佳方法為與淋巴結病理及臨床特徵相互對照;必要時 CXCL13 染色與基因重組研究可有助益。
預後
- 預後不良;初始治療反應佳但常短暫。中位存活 <36 個月,5 年整體存活率介於 30% 與 35% 之間。死亡常因感染併發症。