Angioimmunoblastic T-cell lymphoma
Angioimmunoblastic T-cell lymphoma
Clinical features Angioimmunoblastic T-cell lymphoma (AITL) is a peripheral T-cell lymphoma characterized by systemic disease including generalized lymphadenopathy, hepatosplenomegaly, anemia, and hypergammaglobulinemia.1 The reported male/female ratio is variable, but age of presentation is in the sixth or seventh decades.1–6 The disease is usually in an advanced stage at presentation with generalized lymphadenopathy and frequent hepatosplenomegaly, bone marrow and skin involvement, and systemic symptoms.1–6 Pleural effusions, ascites, arthritis and/or arthralgia, ear, nose, and throat involvement, and neurological manifestations may also be present.1–6 Often, patients have hemolytic (Coombs positive) anemia, polyclonal hypergammaglobulinemia, raised LDH, leukocytosis, including eosinophilia, or lymphopenia and thrombocytopenia.1–4,6 Additional autoimmune phenomena may be encountered including cold agglutinins, cryoglobulins, circulating immune complexes, smooth muscle antibody, rheumatoid factor, and antinuclear antibodies.1,2,4,7
Cases treated with only locally directed therapy, including biopsy or excision alone, almost invariably go into complete remission and, although relapses may occur, no disease related deaths have yet been reported.2–10
The main challenge is therefore to differentiate these lesions from the other lymphomas with a cytotoxic phenotype that often exhibit a more aggressive behavior.11 Clinicopathological correlation should help discriminate from other specific cutaneous lymphomas that occasionally express CD8 rather than CD4 such as mycosis fungoides, pagetoid reticulosis, primary cutaneous anaplastic large cell lymphoma, lymphomatoid papulosis, or others which are also characterized by a cytotoxic phenotype, including CD8 expression in some or all cases. The latter include subcutaneous panniculitis-like T-cell lymphoma, primary cutaneous CD8-positive aggressive epidermotropic cytotoxic T-cell lymphoma, primary cutaneous γ/δ T-cell lymphoma, and extranodal NK/T-cell lymphoma of nasal type. However, the distinction may not always be clear-cut, and localized nonepidermotropic cutaneous CD8-positive lymphoproliferations with a poor outcome have previously been described.12 Expression of CD68 by the neoplastic lymphocytes may help as it has recently been reported to be specific for primary cutaneous acral CD8+ T-cell lymphoma, although the utility of this marker has yet to be tested in large series of cases.13
Cutaneous lesions occur in up to 50% of patients at presentation and/or relapse.6–9 Skin involvement manifests as a pruritic maculopapular eruption on the trunk and extremities; this may mimic a viral exanthem or drug hypersensitivity reaction, and in some cases skin lesions may follow drug administration (Figs 29.141 and 29.142).8–12 Other reported manifestations include urticaria, erythroderma, erosions, petechiae, purpura, papulovesicular prurigo-like lesions, plaques, and tumor nodules.8,13,14
The outlook for patients with AITL is poor, and while the initial response to treatment is good, it tends to be short-lived.1,3,5,6 The median survival is <36 months, and 5-year overall survival is between 30% and 35%.1,3,5,6 Death is often due to infectious complications.3,7,15
Pathogenesis and histologic features AITL is a malignancy of follicular helper T cells, as evidenced by the immunophenotype and gene expression profile.2,3,16–20 Chromosomal
1445 Adult T-cell leukemia/lymphoma
abnormalities have been detected in AITL, the most common being trisomies of chromosomes 3, 5, 18, and 19, gain of chromosome X, and deletion of chromosome 7.2,4,21 Comparative genomic hybridization (CGH) has also documented gains of 11q13, 19, and 22p, and losses of 13q in a subset of cases.22 More recent studies have shown that mutations of epigenetic regulators, such as RHOA, TET2, DNMT3, and IDH2, are common in AITL, although only IDH2 mutations appear to be relatively specific for this subtype of T-cell lymphoma.23–28
Lymph nodes involved by AITL typically show partial effacement of their architecture by a polymorphic infiltrate, mainly in paracortical areas. The sinuses are usually preserved, but perinodal connective tissues are often involved. In the early stages of the disease, there may be hyperplastic B-cell follicles, but frequently follicles are regressed or absent.1–3,29 Paracortical areas show marked proliferation of high endothelial venules and a mixed population of lymphocytes, plasma cells, eosinophils, and histiocytes. The neoplastic lymphocytes are usually of intermediate size with abundant clear or pale cytoplasm.1–3 They are typically found in small clusters around follicles and high endothelial venules. Small reactive lymphocytes are also present, as are scattered large B immunoblasts, some resembling Reed-Sternberg cells.
Biopsies of cutaneous lesions most often show superficial or superficial and deep perivascular or rarely periadnexal lymphocytic infiltrates that are suspicious or diagnostic of lymphoma. The infiltrates range from sparse to dense, contain pleomorphic atypical lymphocytes, and are associated with vascular hyperplasia with prominence of endothelial cells.8,30,31 Less commonly, the changes are non-specific, comprising superficial perivascular infiltrates of small lymphocytes showing no atypia, eosinophils, and sometimes plasma cells.8,30 Mild interface changes have been described in a number of cases in one series.30 Biopsies showing leukocytoclastic vasculitis and granulomatous inflammation are seen in some cases (Figs 29.143 and 29.144).8,32,33 Rarely, the associated and sometimes neoplastic B-cell expansion may dominate the histologic picture, masking the underlying T-cell lymphoma.31,34 Most often, the B-cell proliferation resembles diffuse large B-cell lymphoma (DLBCL), but in others it may mimic lymphoplasmacytic or marginal zone lymphoma, or a plasma cell neoplasm.29,34,34
are highlighted by CD21, CD23, CD35, and/or CNA42. In the skin, it is often harder to identify the neoplastic lymphocytes, but immunohistochemistry with antibodies to CXCL13 seems to be sensitive and specific.30
Clonal rearrangements of the TCR gene are identifiable in lymph nodes in most cases, and correlate well with the presence of CXCL13-positive tumor cells in the skin.11,30,37,38 Clonal immunoglobulin gene rearrangements are seen in 20–30% of cases and correspond to expanded populations of EBV-positive B cells.37,38
Tumor cells often best highlighted in lymph nodes are CD4 positive, and usually express CD2, CD3, and CD5, although significant numbers of reactive CD8-positive T cells may also be present. In common with follicular T-helper cells, at least a proportion express CD10, CXCL13, PD1, and sometimes bcl6.1,2,16,17,19,20,35,36 CD20 highlights the B immunoblasts, and these are often positive for EBV by in situ hybridization. Irregular, expanded FDC meshworks, often in the vicinity of high endothelial venules,
Differentiating cutaneous involvement by AITL from reactive cutaneous infiltrates is best achieved by correlation with the lymph node pathology and clinical features. In certain situations, staining for CXCL13 and gene rearrangement studies may prove useful.30
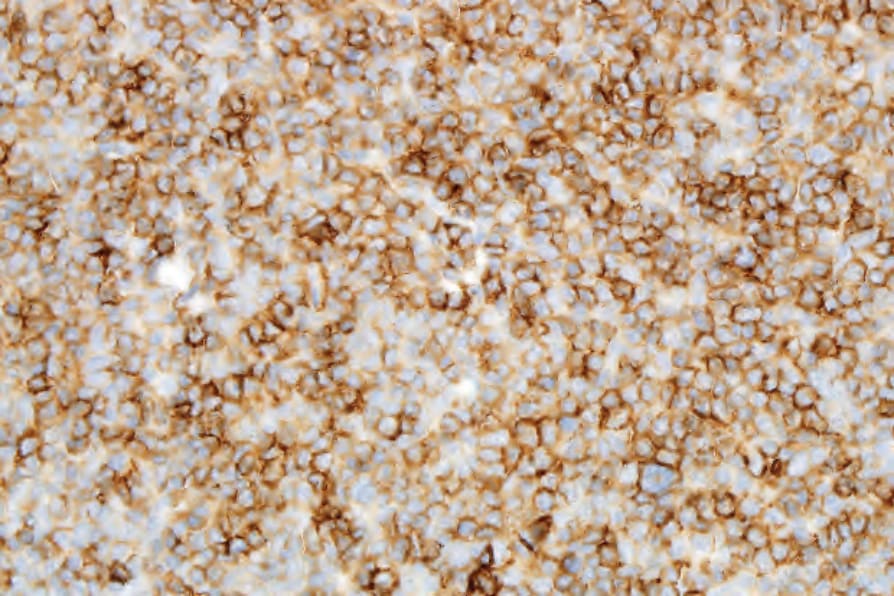
Fig. 29.140 Indolent CD8-positive lymphoid proliferation of the ear: the tumor cells are positive for CD3. Courtesy of Dr. T. Petrella, MD, Dijon, France.
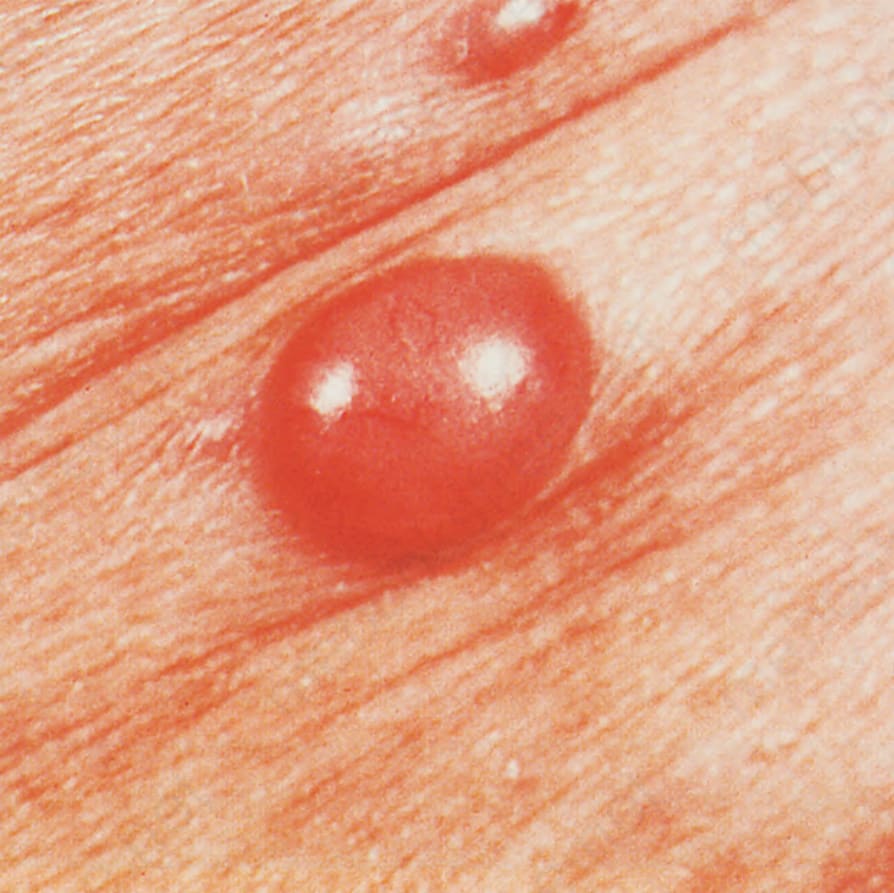
Fig. 29.141 Angioimmunoblastic T-cell lymphoma: an erythematous maculopapular eruption on the anterior chest wall and anterior axillary fold. By courtesy of M.G. Bernengo, MD, Clinica Dermatologica, Turin, Italy.
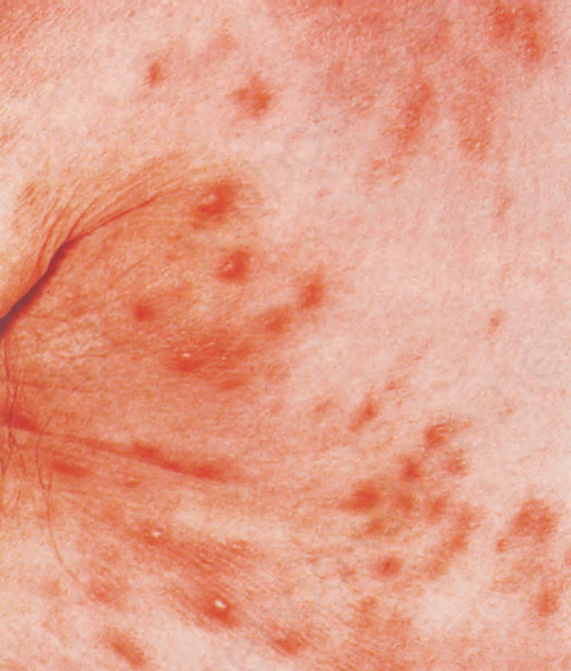
Fig. 29.142 Angioimmunoblastic T-cell lymphoma: a nodular deposit was also present. By courtesy of M.G. Bernengo, MD, Clinica Dermatologica, Turin, Italy.
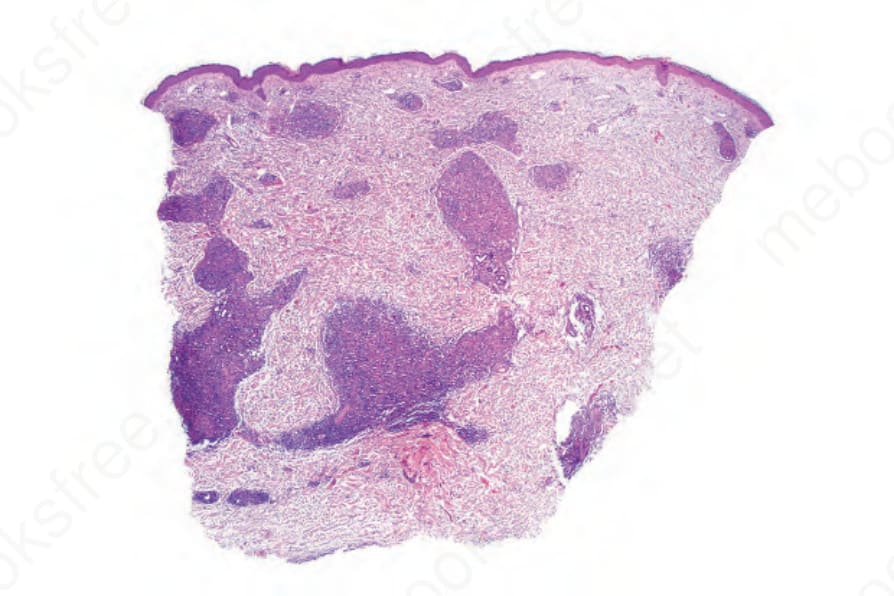
Fig. 29.143 Angioimmunoblastic T-cell lymphoma: this biopsy shows a striking granulomatous dermal infiltrate.
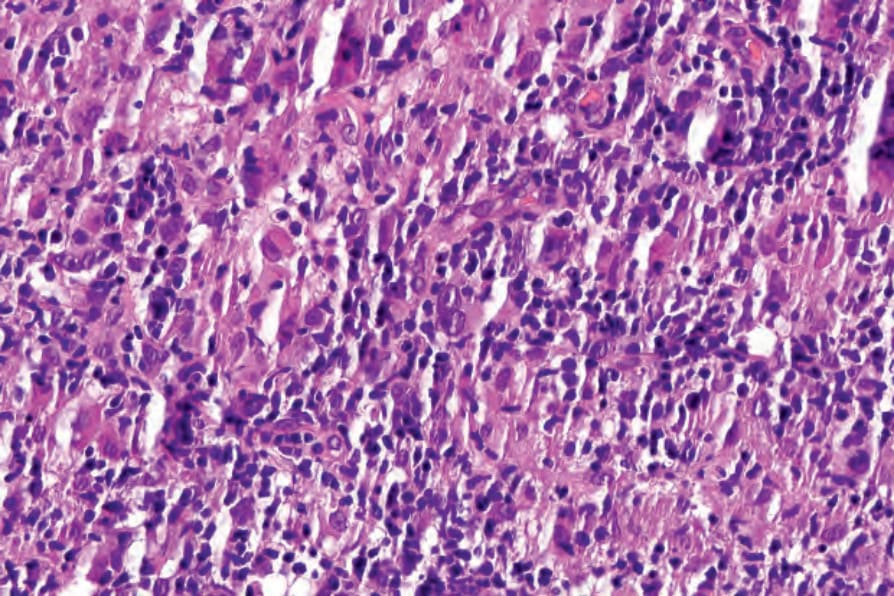
Fig. 29.144 Angioimmunoblastic T-cell lymphoma: note numerous histiocytes and scattered atypical cells.