定義與流行病學
- 蕈狀肉芽腫(mycosis fungoides, MF)雖罕見,卻是最常見的原發性皮膚 T 細胞淋巴瘤(primary cutaneous T-cell lymphoma)。Alibert 於 1806 年依疾病末期出現的蕈樣腫瘤命名。
- 美國年發生率為每 10 萬人 0.36 至 0.46 例,每年約診斷 1000 例新病例;歐洲較少。
- 男性較好發(2:1),黑人較常見(2:1),亞洲人與西班牙裔較少。任何年齡均可,但第四至第六十年(40–60 歲)發生率較高。
- 病程與結果無法預測,從持續、相對良性的疾病,到高罹病率與高死亡率的廣泛性惡性病皆有。
臨床型態與分期
- 除典型(Alibert)型外,可呈 poikilodermatous 變異(poikiloderma atrophicans vasculare、large plaque parapsoriasis)或紅皮症(Hallopeau-Besnier)。後者不應與 Sézary syndrome 混淆。
- tumeur d’emblée mycosis fungoides(Vidal Brocq)已不再視為一個獨立病種,這類病例多屬其他 T 細胞淋巴瘤變異。
- 較罕見表現包括 bullous、follicular、hypopigmented、verrucous/hyperkeratotic、pustular、lichenoid papular、palmoplantar psoriasiform、granulomatous 與 acanthosis nigricans 樣變異,病程與典型 MF 相似,不視為獨立病種。
- folliculotropic MF、pagetoid reticulosis、granulomatous slack skin disease 則具獨特臨床病理特徵,在 WHO 分類中被認定為 MF 的生物學上不同變異。
- 典型 MF 傳統分為 patch、plaque、tumor 三期,但此分類略嫌武斷,因三期可同時出現於同一人,部分病人則永不超過 patch 期。
- 早期紅斑病灶為不規則、不對稱、輕微脫屑、大小不一的粉紅或紅色斑塊,多見於軀幹、肢帶部、乳房與屈側。
- 臨床鑑別包括 discoid/atopic/contact allergic dermatitis、psoriasis,尤其是 chronic superficial scaly dermatitis(small plaque parapsoriasis,又稱 digitate dermatosis);後者病灶大小、形狀、顏色一致,與 MF 多變者形成鮮明對比。
- 藥物不良反應亦可擬似 MF,常見藥物包括抗痙攣劑(phenytoin、barbiturates、carbamazepine)、心臟用藥(atenolol、ACE inhibitors)、抗組織胺、ciclosporin 與 allopurinol。
分期與預後
- 臨床分期採 ISCL 與 EORTC 系統,評估皮膚病灶範圍與性質、淋巴結侵犯、內臟侵犯及周邊血液侵犯程度(Table 29.3)。
- 僅 patch/plaque 且覆蓋 ≤10% 體表面積(T1)時,存活率與年齡匹配對照組無異。
- patch/plaque 覆蓋 >10% 體表面積(T2)者中位存活 10–12 年,進展風險 25%;有腫瘤(T3)或紅皮症(T4)者中位存活僅 4–5 年。
- 內臟侵犯預後極差,中位存活僅 1–2 年。淋巴結腫大常見,但不必然代表病理侵犯。
- 不良預後指標:lactate dehydrogenase (LDH)、soluble IL-2 receptor、ESR 上升、血嗜酸性球增高,以及向 large cell lymphoma 轉化。
- 感染為主要死因,最常見病原為 Staphylococcus aureus、Enterobacteriaceae 與 Pseudomonas aeruginosa。
- MF 病人罹上皮腫瘤(肺癌、結腸癌)與 B 細胞 non-Hodgkin lymphoma 風險增加。
致病機轉
- MF 自始即為單株 T 細胞的腫瘤性增生。clonal TCR gene rearrangement 雖在多種非腫瘤情況亦可見,但高度提示淋巴瘤過程,尤其同一株出現於多處病灶或長期持續時。
- 病因與致病機轉可能為多因子,特定致病原與精確機轉未知;遺傳易感性證據少,家族性發生極罕見。
- 反覆性染色體異常包括 1p、17p、10q/10、13q、19 缺失,以及 4/4q、17q/17、18 增多。
- 多認為 MF 由慢性抗原刺激(長期接觸刺激物或感染原)發展而來;流行病學指向職業性接觸金屬、塑膠、切削油、溶劑等。感染原(HTLV-I、HTLV-II、EBV、CMV、HHV6/7/8)研究結果矛盾或不確定。
- 腫瘤抑制與凋亡相關基因(p15、p16、Nav3、PTEN、p53)的沉默與 plaque 至 tumor 期進展有關。大規模平行定序發現表觀遺傳調控、T 細胞受體訊息、特別是 JAK-STAT 與 NF-κB 路徑的反覆突變。
- 腫瘤細胞表現 CCR4 與 CCR10;其配體 CCL17、CCL22(CCR4)與 CCL27(CCR10)在病灶高量表現。腫瘤細胞偏向 Th2 表型,表現 IL-4、IL-5、IL-18,並具抗凋亡特性(Fas 基因突變、TNF 訊息路徑異常)。
組織病理
- 表皮趨向性(epidermotropism)為 MF 的組織學標誌,與疾病期別及淋巴球分化程度相關;於 patch 與 plaque 期較明顯,tumor 期可完全喪失。
- 與濕疹的淋巴球外滲不同,MF 的 epidermotropism 通常無或僅極輕度海綿水腫,且通常不見水疱形成。
- 表皮內含異型淋巴細胞的 Pautrier microabscess 為 MF 典型但非診斷性特徵。
- 大型細胞具高度不規則、捲曲或腦回狀(cerebriform)細胞核,稱 Sézary 或 mycosis 細胞;電顯下為多葉、高度捲曲核並具明顯周邊染色質邊集。
Patch 期
- 早期病理特徵常細微而易被忽略,常需多次切片。表皮厚度可正常、輕度棘層肥厚或較少見的萎縮,常有輕度過度角化伴局灶角化不全。
- 特徵為表皮內少數異型不規則淋巴細胞,各圍以透明暈(halo);基底層沿線之淋巴球柵狀排列為診斷指標;個別壞死角質細胞可見。淋巴組織球浸潤環繞淺層血管叢。
- 先前以 steroids 或 PUVA 治療可能遮蔽特徵,強調臨床病理對照之必要。
Plaque 期
- 已確立的 plaque 期通常診斷不難。有緻密過度角化、斑狀角化不全,表皮常呈棘層肥厚並常呈 psoriasiform 外觀。
- 浸潤較 patch 期強烈,表皮內常見大量異型單核細胞;真皮分布以淺層帶狀為主。
- Pautrier microabscess 不罕見,見於 17–37.5% 病例。可累及毛囊上皮(有時伴 follicular mucinosis)與汗管上皮(syringotropism)。
- poikilodermatous 病灶表皮典型扁平、萎縮,伴基底層水樣變性與真皮帶狀/血管周圍浸潤、色素失禁與微血管擴張。
Tumor 期
- 極緻密浸潤占據真皮,有時延伸入皮下脂肪,可呈 top-heavy 構型;病灶常潰瘍,epidermotropism 通常輕微或缺如。
- 浸潤含大量高度多形性細胞,有絲分裂明顯且常異常。
免疫表型與分子
- MF 多為 CD4+、CD45RO+ helper/memory T 淋巴球,偶見 CD8+ 甚至 CD4/CD8 表型(對預後無影響)。
- 通常表現泛 T 細胞抗原 CD2、CD3、CD5、CD7 及 TCRαβ、CLA。CD7 常早期局灶喪失,但無診斷價值(反應性病變亦常喪失)。
- CD25(IL-2 receptor)在多達 50% 病例陽性;PD1 常表現。CD30 表現與轉化相關但無預後意義。MUM1 罕見表現。偶見異常 CD20 表現。
- 多數病例可由 Southern blot 或 PCR 檢出 clonal TCR gene rearrangement:tumor 期可達 100%、plaque 期 50–100%、patch 期 50–78%。但需謹慎判讀,因多種發炎性皮膚病(discoid lupus erythematosus、lichen planus、lichen sclerosus、PLEVA)亦有此重排。
淋巴結侵犯(dermatopathic lymphadenopathy)
- MF 常有淋巴結病變,但不必然對應淋巴瘤性侵犯。dermatopathic lymphadenitis 特徵為結內副皮質區大量組織球(含 interdigitating reticulum cells 與 Langerhans cells)浸潤,組織球常含黑色素,此階段淋巴結結構未變形。
- 進展時副皮質可見 mycosis/Sézary 細胞浸潤,最易於 postcapillary venule 周圍辨識;晚期出現腫瘤細胞片狀、淋巴結結構消失。
- 兩個歷史分期系統(NCI-VA 與 Dutch system)對「異常」淋巴球定義不同(Dutch 僅接受 >7.5 微米的 cerebriform 細胞為腫瘤性),均已納入 ISCL/EORTC 簡化版(Table 29.4)。
鑑別診斷
- patch 期 MF 須與 chronic superficial dermatitis 區分(後者為輕度海綿樣過程,無淋巴球異型與 epidermotropism)。
- Lymphomatoid drug reactions 在組織學上可能與 MF 無法區分。
- 顯著 epidermotropism 的 MF 須與 CD8+ primary cutaneous epidermotropic T-cell lymphoma 區分;ATLL 常呈與 MF 無法區別的組織學。
- ISCL 提出加權評分演算法(最高 6 分,診斷 MF 至少需 4 分)。
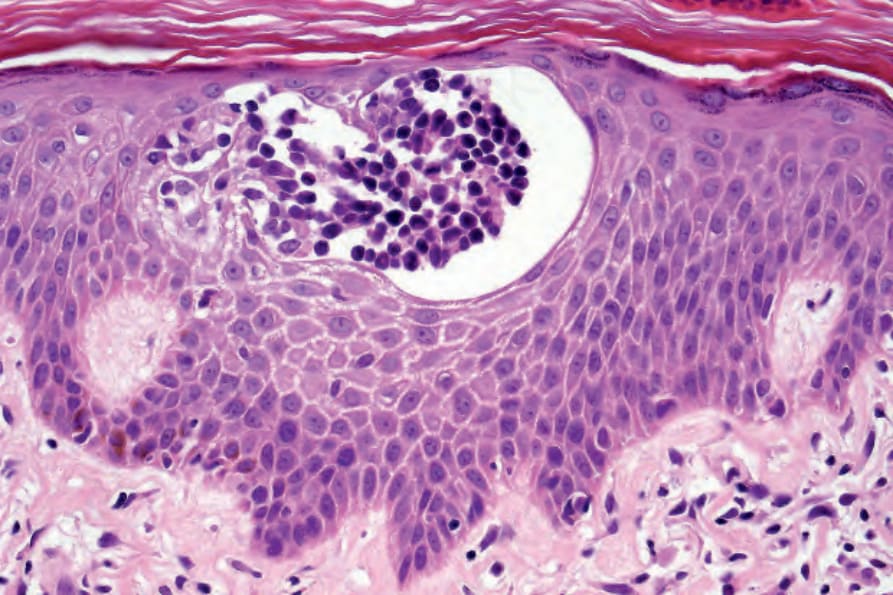
圖 29-17:Mycosis fungoides 典型的 Pautrier microabscess,由染色深的異型淋巴細胞組成。
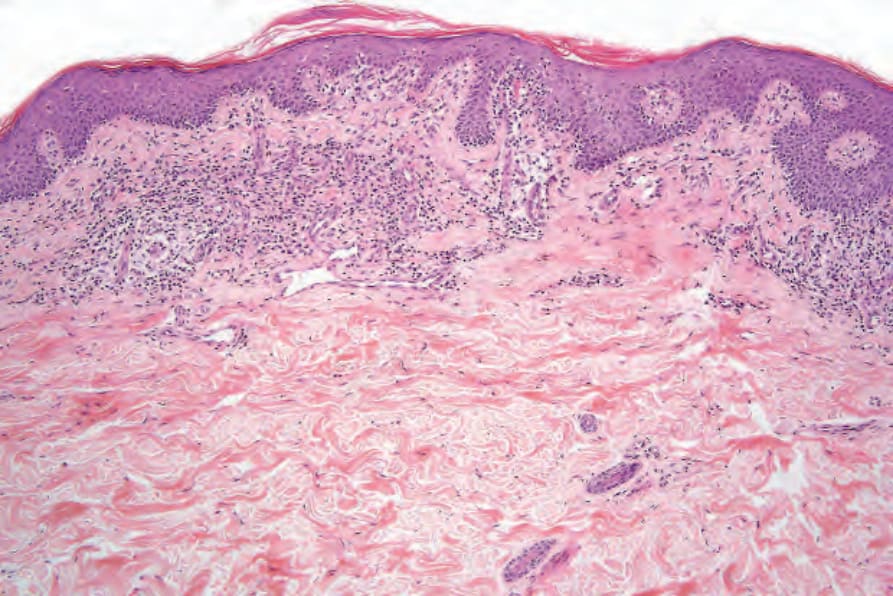
圖 29-18:Mycosis fungoides(patch 期)早期病灶切片,顯示局灶角化不全、棘層肥厚、異型淋巴細胞沿真皮表皮交界「貼附」及淺層血管周圍淋巴組織球浸潤。
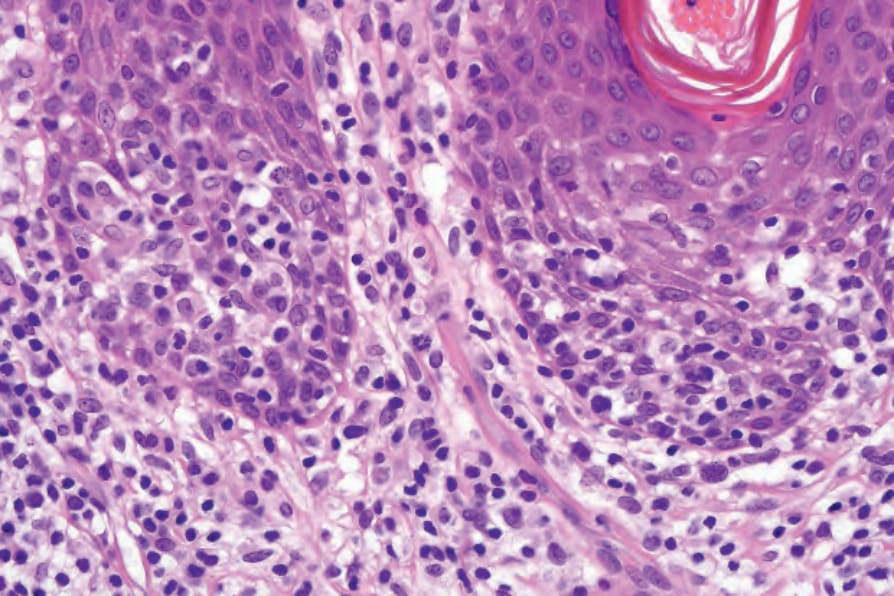
圖 29-29:Mycosis fungoides(plaque 期)緻密的異型淋巴細胞群並有顯著 epidermotropism。