Mycosis fungoides
Mycosis fungoides
Clinical features Mycosis fungoides (MF) (Gr. mykes, fungus; L. fungus + Gr. eidos, form), although rare, represents the commonest form of primary cutaneous T-cell lymphoma.1–7 Alibert named it, in 1806, after the mushroom-like tumors that develop in the terminal stages of the illness (Fig. 29.1). The annual incidence in the United States varies from 0.36 to 0.46 cases per 105 of the population,8–10 with approximately 1000 new cases diagnosed per year.1,11 The incidence in Europe is somewhat less.12 There is predilection for males (2 : 1). It is more common in blacks (2 : 1) and less common in Asians and Hispanics.2,8,11,13 Any age group may be involved, but there is a higher incidence in the fourth to sixth decades. MF in children is discussed separately (see below).
The course and outcome is unpredictable, ranging from a protracted, persistent, relatively benign illness through to a widespread malignancy with high morbidity and mortality.3
In addition to the classical (Alibert) form, patients may present with a poikilodermatous variant (poikiloderma atrophicans vasculare, large plaque parapsoriasis) or with erythroderma (Hallopeau-Besnier). The last should not be confused with Sézary syndrome, which represents an erythrodermic leukemic manifestation of T-cell lymphoma, and usually develops de novo.
1406 Cutaneous lymphoproliferative diseases and related disorders
MF presenting with tumor nodules from the outset, so-called tumeur d’emblée mycosis fungoides (Vidal Brocq), is no longer recognized as an entity. Such cases are likely to represent other variants of T-cell lymphoma including cutaneous anaplastic large cell lymphoma or primary cutaneous T-cell lymphoma, unspecified (CD30-negative large cell lymphoma).
Rarer presentations include bullous, follicular, hypopigmented, verrucous/ hyperkeratotic, pustular, lichenoid papular, palmoplantar psoriasiform, granulomatous, and acanthosis nigricans-like variants. These are clinically unusual cases that run a similar course to that of classic MF, and are not considered separate entities. In contrast, folliculotropic mycosis fungoides, pagetoid reticulosis, and granulomatous slack skin disease have distinct clinicopathological features and are recognized as biologically distinct variants of MF in the WHO classification.14,15
Classic MF is traditionally divided into patch, plaque, and tumor stages.2 This is, however, a somewhat arbitrary classification because all stages may be present simultaneously in one individual while other patients never progress beyond the patch stage.15,16 In addition, patch stage lesions obviously merge with plaques.
The early erythematous lesions are irregular, asymmetrical, slightly scaly, variably sized pink or red patches (Fig. 29.2). Many lesions show signs of atrophy, and in some lighting conditions they appear smooth and shiny. While lesions are more commonly present on the trunk, limb girdles, breasts, and flexures; they can also be more widespread (Figs 29.3 and 29.4).
The clinical differential diagnosis includes discoid, atopic or contact allergic dermatitis, psoriasis, and, in particular, chronic superficial scaly dermatitis (small plaque parapsoriasis).17–22 Patients, usually middle aged, present with erythematous scaly persistent patches, showing predilection for the limbs and trunk, that are sometimes likened to cigarette paper.20,23 While the lesions may be round or oval, they often have a finger-like appearance – hence, the alternative designation digitate dermatosis (Fig. 29.5). The patches tend to be uniform in size, shape, and color, contrasting vividly with the great variability of those of mycosis fungoides.
Adverse drug reactions may also mimic mycosis fungoides. Patients can present with multiple infiltrated plaques or erythroderma that are histologically indistinguishable from mycosis fungoides.22 Drugs which have been particularly implicated are the anticonvulsants (including phenytoin, barbiturates, carbamazepine), cardiac drugs such as atenolol and angiotensin-converting enzyme (ACE) inhibitors, antihistamines, ciclosporin, and allopurinol.22,24–26
Patients sometimes develop foci of poikiloderma within a more typical background. In others, the entire eruption may be poikilodermatous – so-called poikiloderma atrophicans vasculare (Gr. poikilos, spotted, mottled, varied) (large plaque parapsoriasis).17 They present with small numbers
of large plaques showing a predilection for the breasts, buttocks, hips, abdomen, and major flexures (Fig. 29.6). Individual features of the plaques include atrophy, telangiectases, and variable hypo- and hyperpigmentation with erythema (Fig. 29.7). The appearances have been likened to those of chronic radiation damage. Further progression may be similar to that of classic mycosis fungoides, although it appears that fewer patients develop tumor-stage mycosis fungoides.27
Further progression of patch stage disease leads to the development of an increased number of indurated plaques (Figs 29.8 and 29.9). The lesions can be quite bizarre and, not uncommonly, due to central regression, they have an annular or serpiginous appearance (Fig. 29.10). Plaques are sometimes extremely hyperkeratotic, particularly on the palms and soles (see mycosis fungoides palmaris et plantaris, page 1421).
1407 Mycosis fungoides
A small proportion of patients develop tumors (Figs 29.11–29.13). Sometimes ulcerative lesions are seen (Fig. 29.14). The face, scalp, and intertriginous areas are particularly affected.28
Clinical staging relates to the extent of disease and the presence or absence of cutaneous tumor nodules and erythroderma. It is based on the system proposed by the International Society for Cutaneous Lymphomas (ISCL) and the EORTC.29 This scheme takes account of advances in molecular biology, immunohistochemistry, and imaging, as well as new data on prognostic variables. It assesses the extent and nature of the skin lesions present, extent of nodal involvement, visceral involvement, and the presence and degree of any peripheral blood involvement (Table 29.3). Combinations of these parameters (stage) can be used to determine prognosis and
are essential for determining treatment.30,31 Outcome in mycosis fungoides is varied. When only patches and plaques are present at presentation, and cover 10% of body surface area (T1), survival is no different from that of age-matched controls.17,32–34 Patients with patches and plaques covering >10% body surface area (T2) have a median survival of 10–12 years and a 25% risk of progression, whilst the median survival for patients with tumors (T3) or erythroderma (T4) is only 4–5 years.33,34 Visceral involvement carries a very poor prognosis, with median survivals of only 1–2 years.31,34 Lymph node enlargement is common and does not necessarily indicate pathological involvement. Its impact on prognosis is often overshadowed by the extent of skin lesions. The presence of abnormal circulating cells and the tumor burden in the peripheral blood are also important parameters to assess as
1408 Cutaneous lymphoproliferative diseases and related disorders
they are of independent prognostic significance.33,35,36 The patient should therefore be thoroughly investigated to assess the extent of skin lesions and to determine whether there is nodal, visceral, or hematological spread.
Raised levels of lactate dehydrogenase (LDH), soluble interleukin (IL)-2 receptor, erythrocyte sedimentation rate (ESR), and raised blood eosinophil count are other markers of poor prognosis.37–43 Transformation to large cell lymphoma also relates to adverse outcome.
Infection is the major cause of death, with Staphylococcus aureus, Enterobacteriaceae and Pseudomonas aeruginosa being the most frequent pathogens.1,2,44–46 Beta-hemolytic streptococcus, herpes simplex, and varicella-zoster infections are also of importance.45
Patients with mycosis fungoides also have an increased risk of epithelial tumors, including carcinoma of the lung and colon, and B-cell non-Hodgkin lymphoma (NHL).47–49
Pathogenesis and histologic features Mycosis fungoides is a neoplastic proliferation of monoclonal T cells from the outset.50–54 While monoclonal lymphoid populations have been described in apparently non-neoplastic conditions, such as pityriasis lichenoides acuta, lichen aureus, lichen planus, pigmented purpuric dermatosis, allergic contact dermatitis, and drug reactions, the presence of a clonal TCR gene rearrangement is highly suggestive of a lymphomatous process, particularly when the same clone is found in more than one lesion or in one patient over time.54–65 Furthermore, recurrent chromosomal abnormalities have been found in some cases including loss of chromosomes 1p, 17p, 10q/10, 13q, and 19, as well as gains of 4/4q, 17q/17, and 18.66
1409 Mycosis fungoides
Stage Description TNMB
IA Limited patches, papules and/or plaques covering <10% skin surface area (T1); no evidence of nodal (No), visceral (M0) or significant blood involvement (B0), or low blood tumor burden (B1)
T1N0M0B0,1
B Generalized patches, papules and/or plaques covering ≥10% skin surface area (T2); no evidence of nodal (No), visceral (M0) or significant blood involvement (B0), or low blood tumor burden (B1)
T2N0M0B0,1
II Any extent of patches, papules and/or plaques (T! 0r T2); clinically abnormal nodes showing DL (N1) or early involvement by MF (N2); no evidence of visceral (M0) or significant blood involvement (B0), or low blood tumor burden (B1)
T1,2N1,2M0B0,1
IIB One or more tumors (≥1.5 cm diameter) (T3); up to early lymph node involvement (N2); no evidence of visceral (M0) or significant blood involvement (B0), or low blood tumor burden (B1)
T3N0–2M0B0,1
III Erythema covering ≥80% skin surface area; up to early lymph node involvement (N2); no evidence of visceral (M0) or significant blood involvement (B0), or low blood tumor burden (B1)
T4N0–2M0B0,1
IIIA Stage III with no significant blood involvement T4N0–2M0B0
IIIB Stage III with low blood tumor burden T4N0–2M0B1
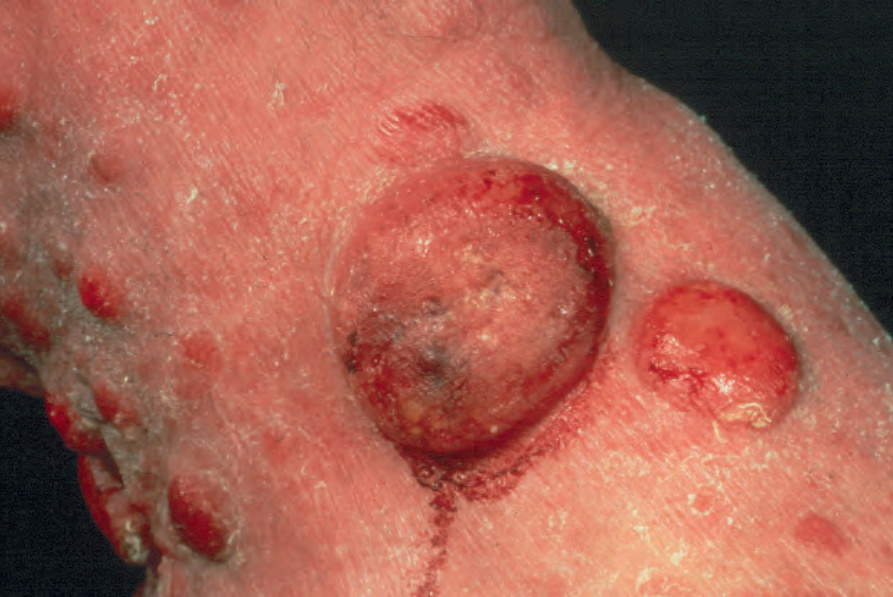
Fig. 29.1 Mycosis fungoides: this patient has advanced tumor-stage mycosis fungoides. Such massive ulcerated tumor nodules are a rare manifestation. By courtesy of the Institute of Dermatology London, UK.
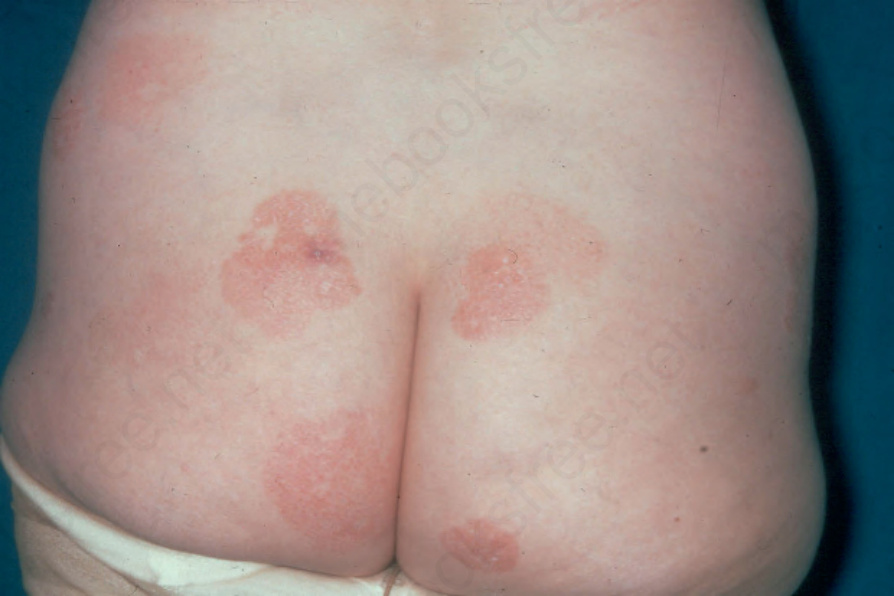
Fig. 29.2 Mycosis fungoides (patch stage): these irregular, erythematous, and scaling lesions are present in a typical distribution on the buttocks. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
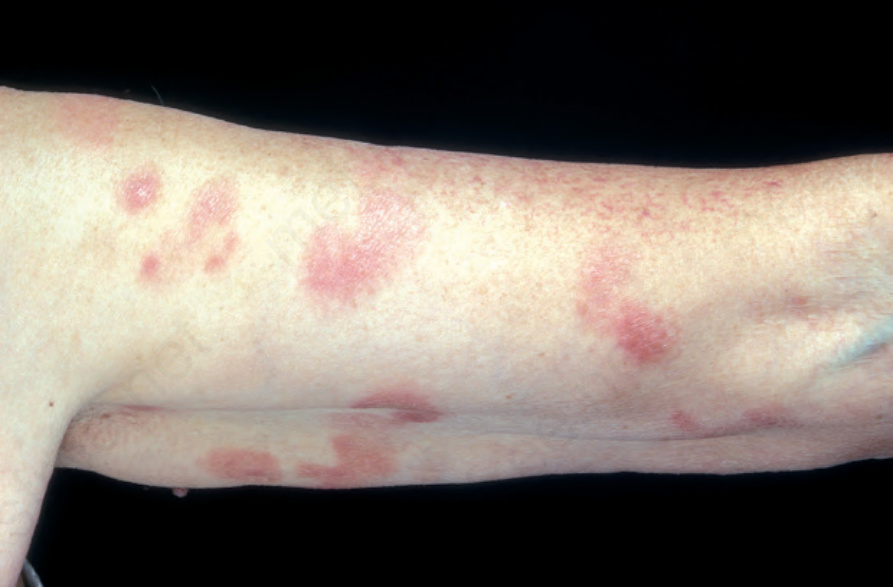
Fig. 29.3 Mycosis fungoides: multiple patches are present on this patient’s arm. By courtesy of the Institute of Dermatology, London, UK.
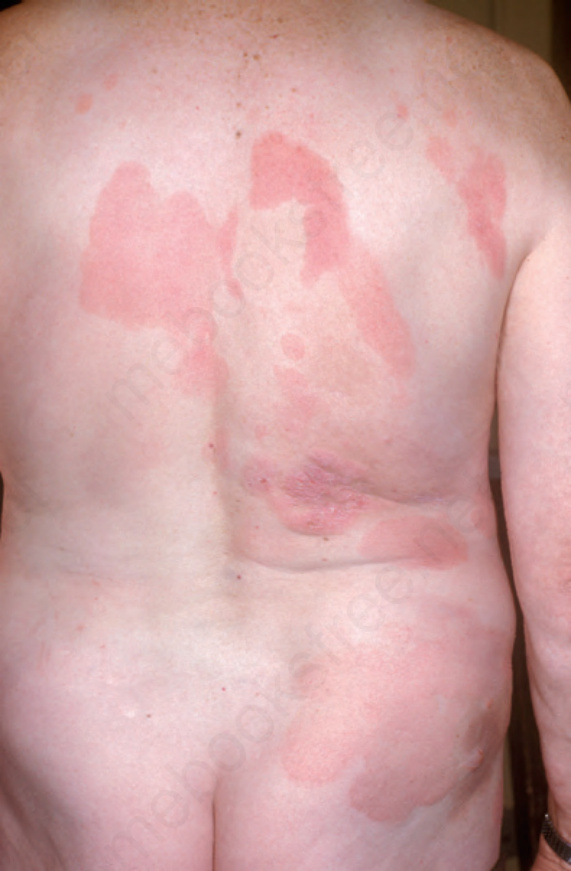
Fig. 29.4 Mycosis fungoides: lesions sometimes have a generalized distribution. By courtesy of the Institute of Dermatology, London, UK.
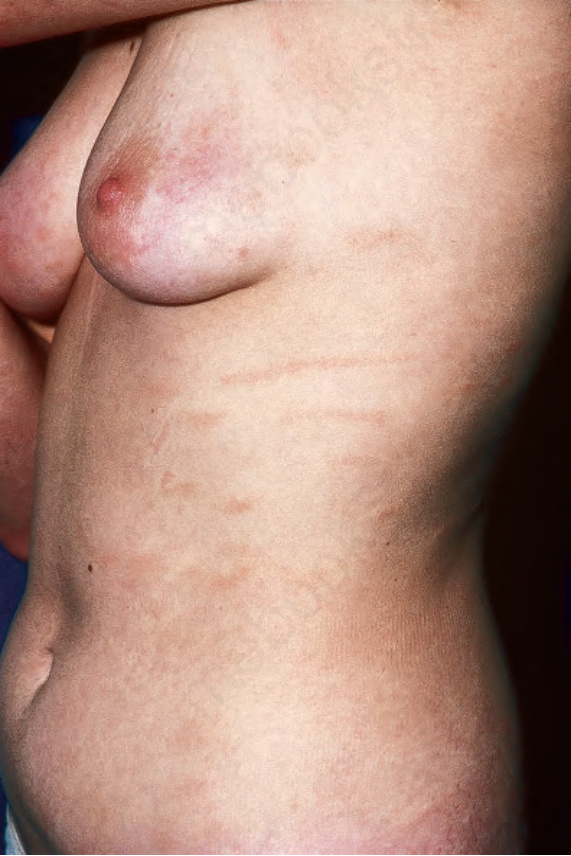
Fig. 29.5 Superficial scaly dermatitis: this patient shows digitate erythematous lesions in a characteristic distribution. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
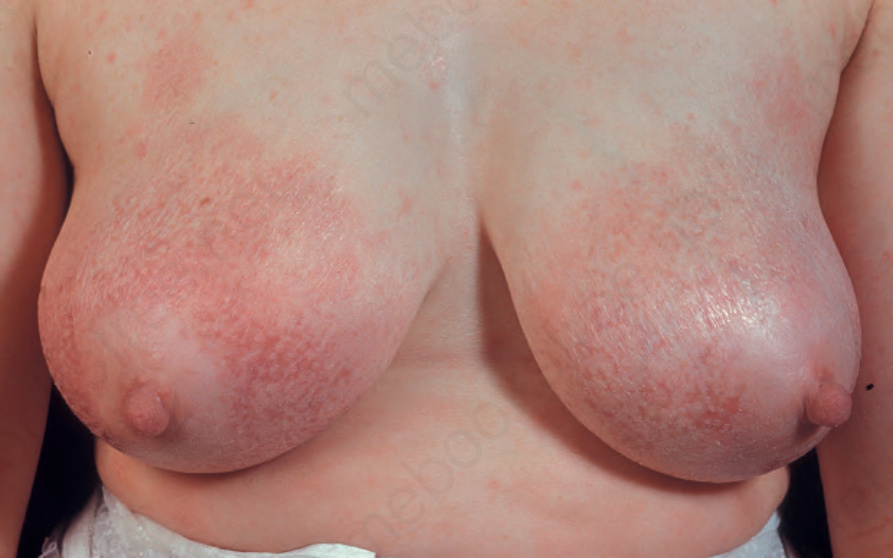
Fig. 29.6 Mycosis fungoides (poikiloderma atrophicans vasculare): early lesion showing slight scaling and dilated vasculature. By courtesy of the Institute of Dermatology, London, UK.
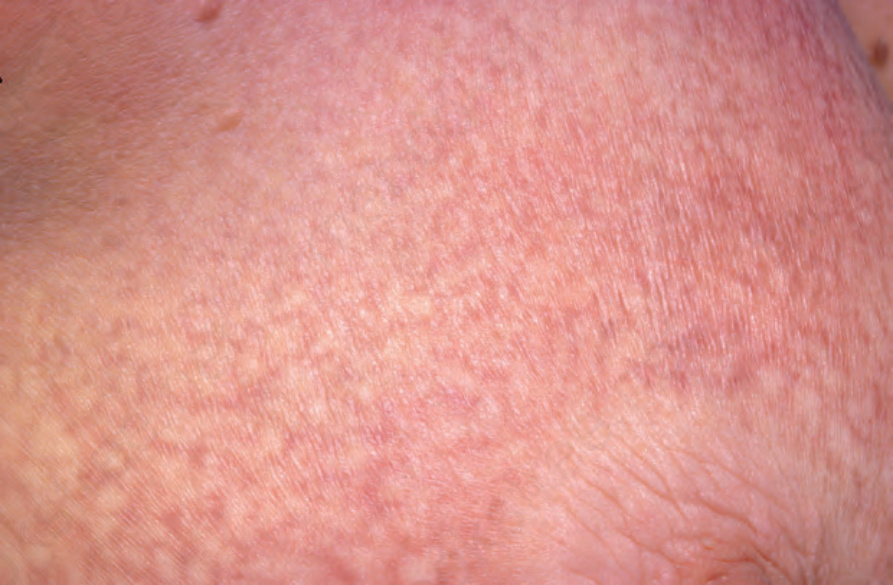
Fig. 29.7 Mycosis fungoides (poikiloderma atrophicans vasculare): this field shows the typical features of reticulate pigmentation, atrophy, scaling, and telangiectasia. By courtesy of the Radcliffe Infirmary, Oxford, UK.
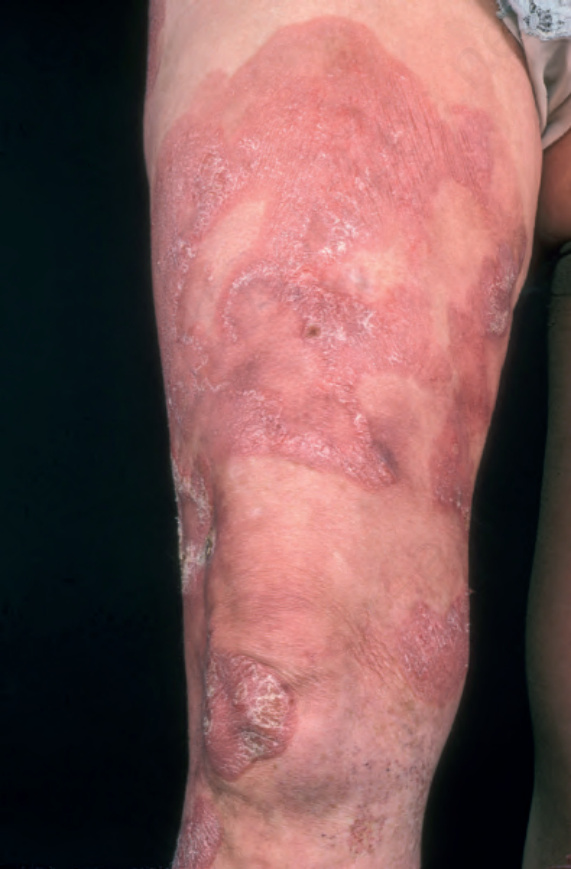
Fig. 29.8 Mycosis fungoides: large erythematous plaques with scaling. By courtesy of the Institute of Dermatology, London, UK.
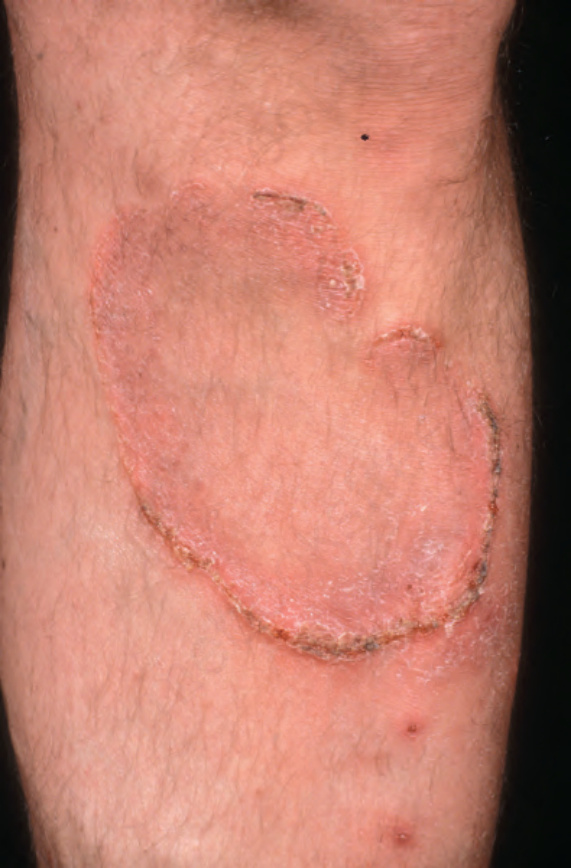
Fig. 29.9 Mycosis fungoides: high-power view. By courtesy of the Institute of Dermatology, London, UK.
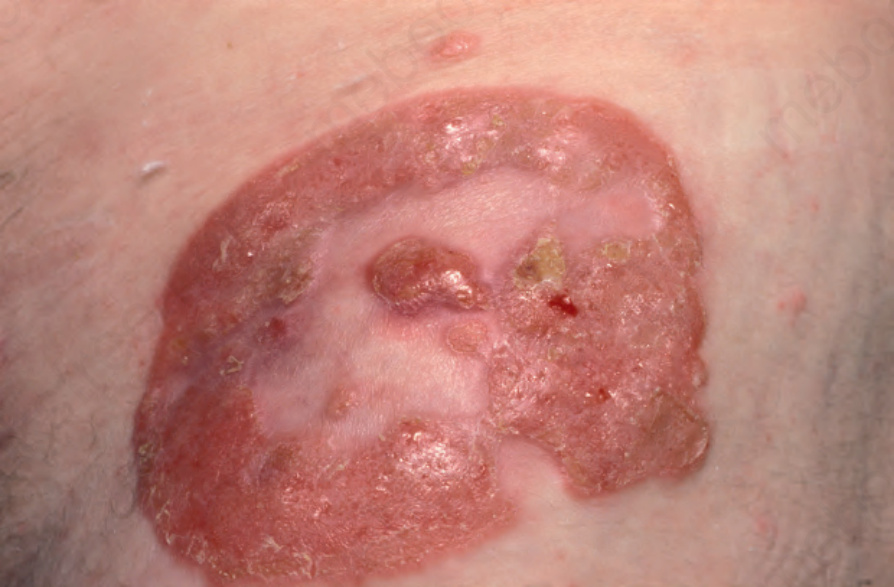
Fig. 29.10 Mycosis fungoides: close-up view of an annular lesion. By courtesy of the late N.P. Smith, MD, Institute of Dermatology, London, UK.
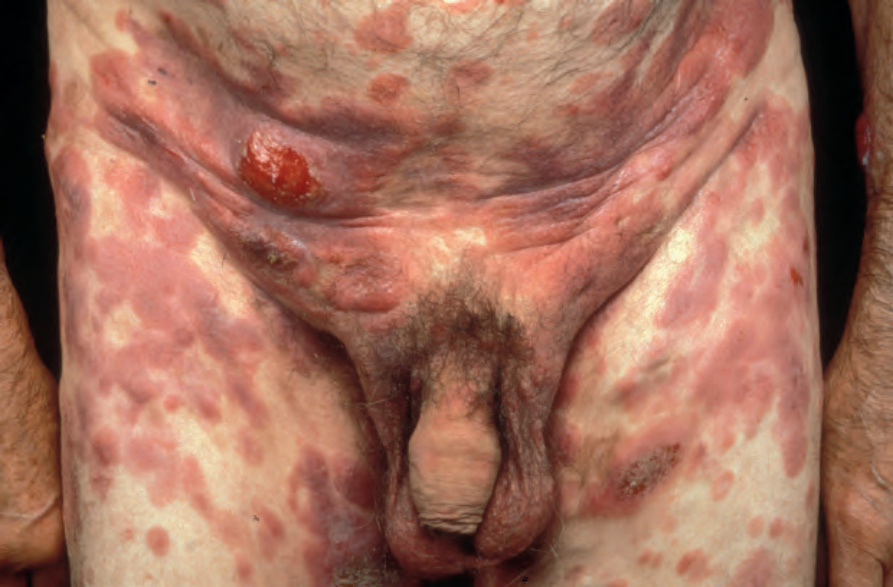
Fig. 29.11 Mycosis fungoides: multiple tumors have arisen against a background of plaque stage disease. By courtesy of the Institute of Dermatology, London, UK.
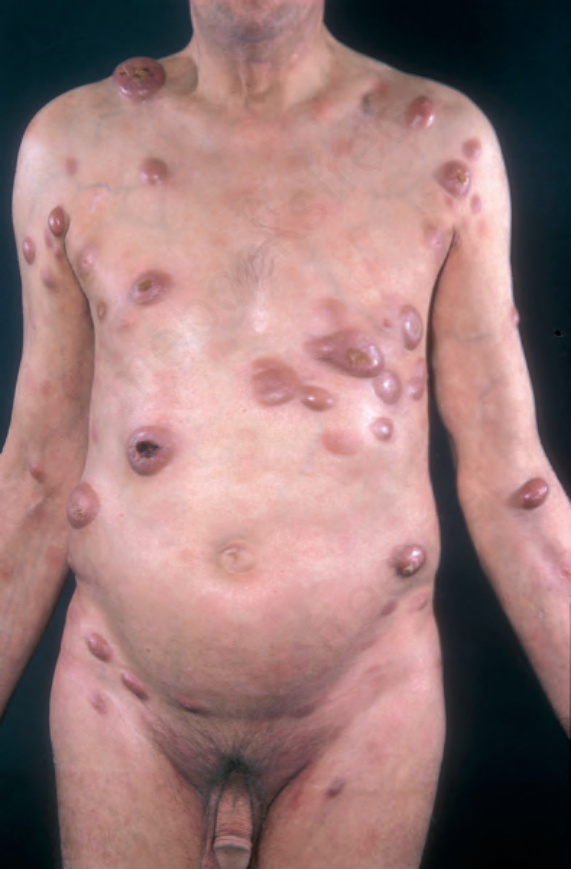
Fig. 29.12 Mycosis fungoides (tumor stage): there are multiple tumor nodules in a background of patches and plaques. By courtesy of the Institute of Dermatology, London, UK.
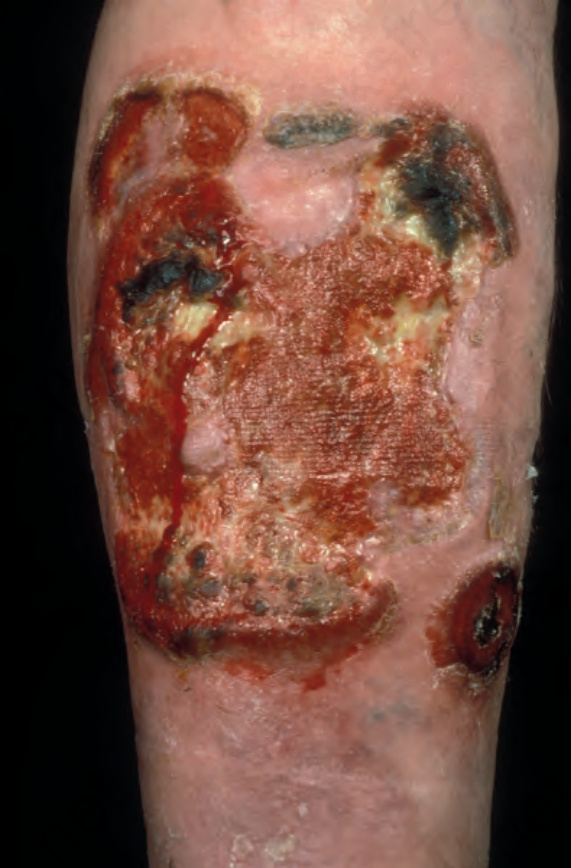
Fig. 29.13 Mycosis fungoides: this patient shows advanced disease with a fungating tumor on the knee. By courtesy of the Institute of Dermatology, London, UK.
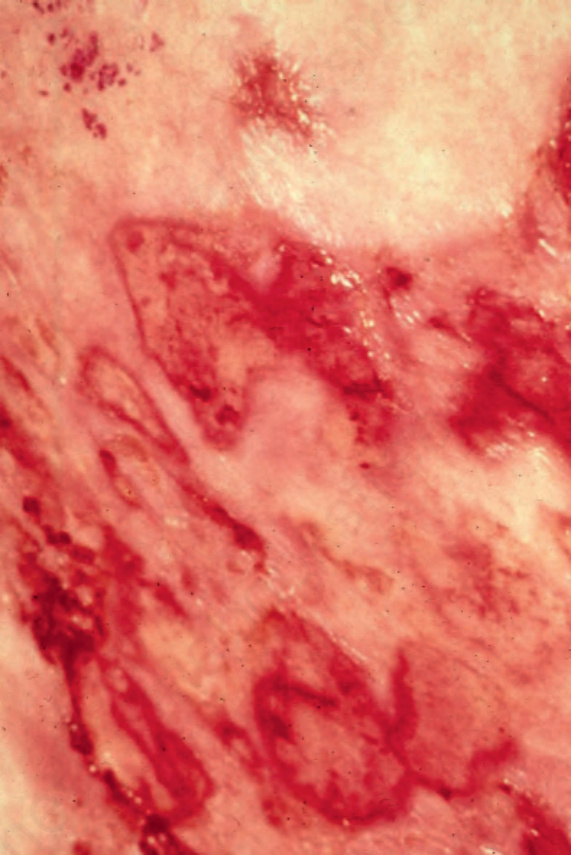
Fig. 29.14 Mycosis fungoides: ulcerative lesions, as seen in this patient, are a rare manifestation. By courtesy of A. du Vivier, MD, King’s College Hospital, London, UK.
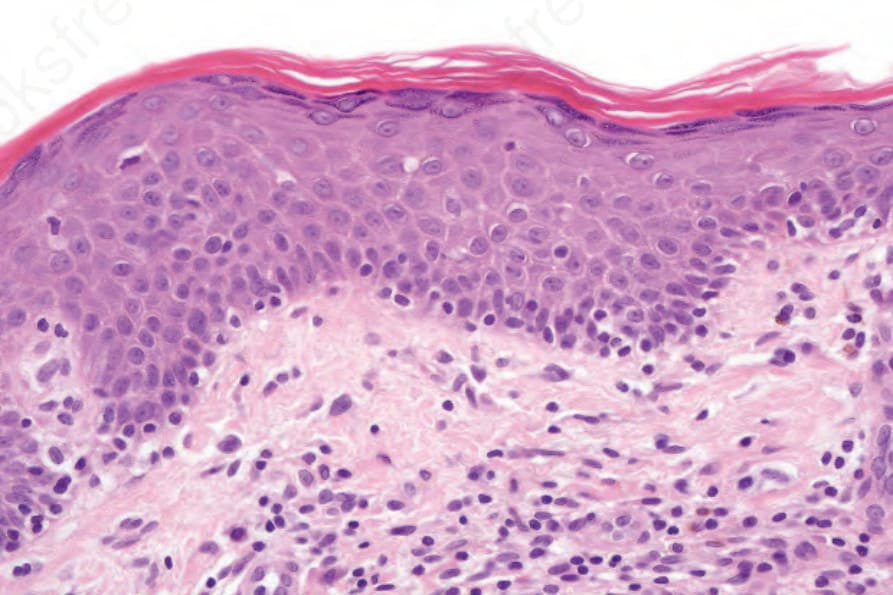
Fig. 29.19 Mycosis fungoides (patch stage): high-power view of atypical lymphocytes tagging the dermal–epidermal junction.
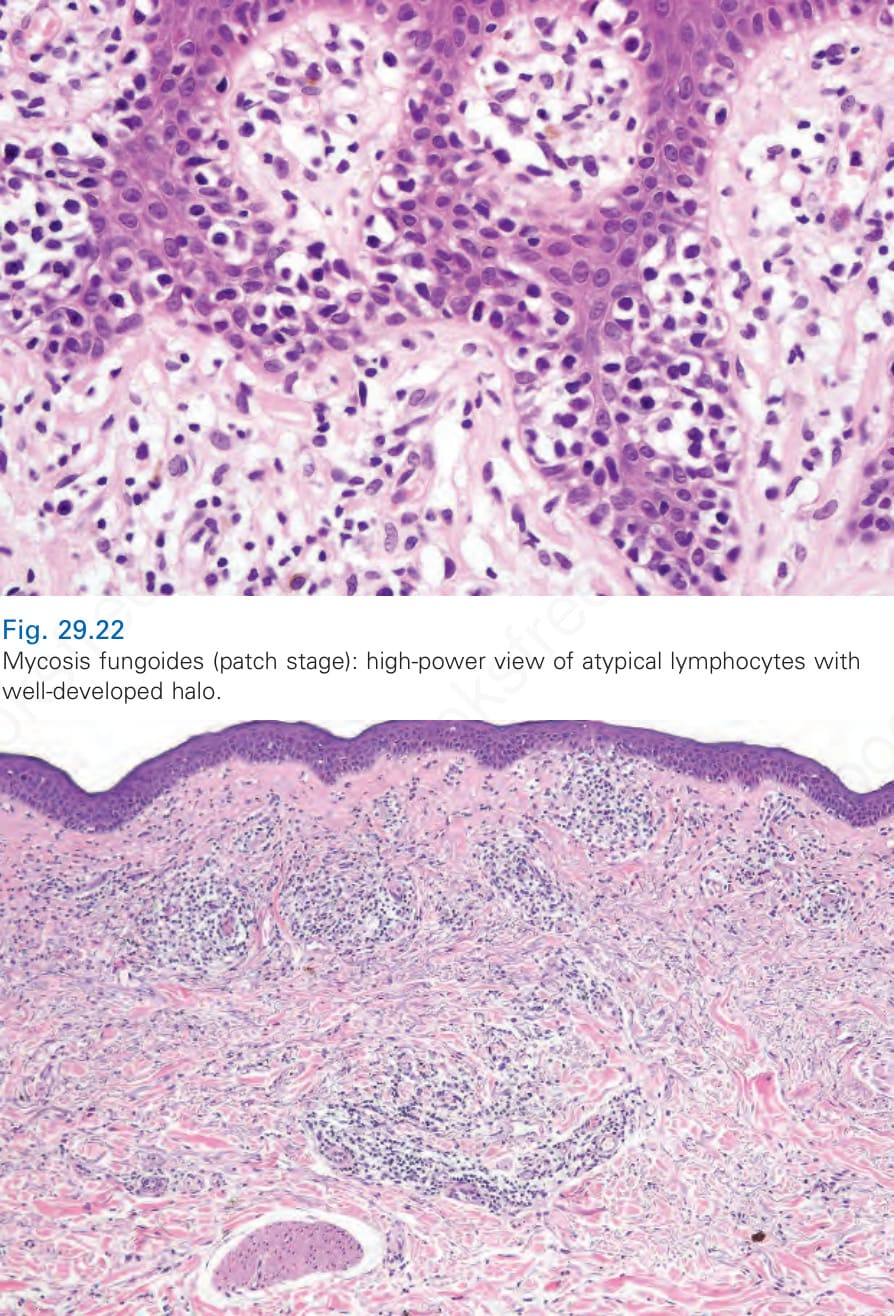
Fig. 29.22 Mycosis fungoides (patch stage): high-power view of atypical lymphocytes with well-developed halo.
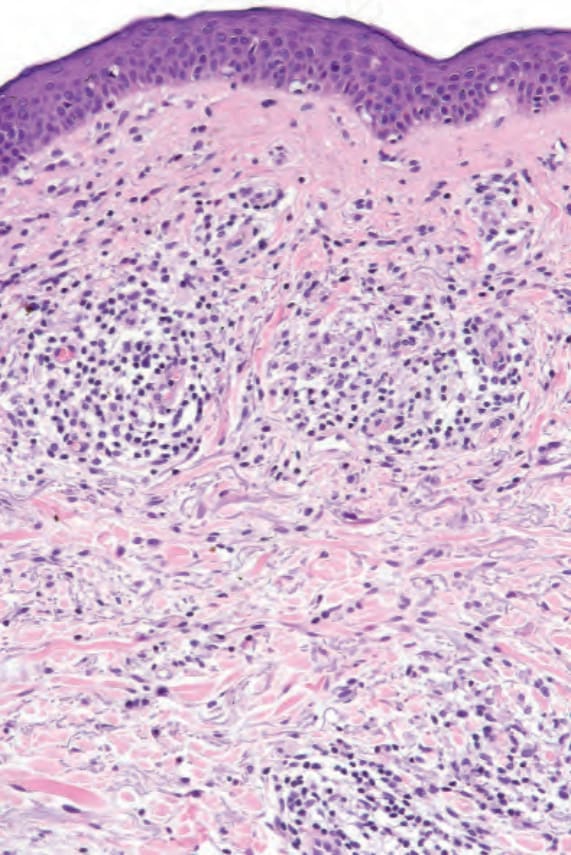
Fig. 29.24 Mycosis fungoides: higher-power view of Fig. 29.23.
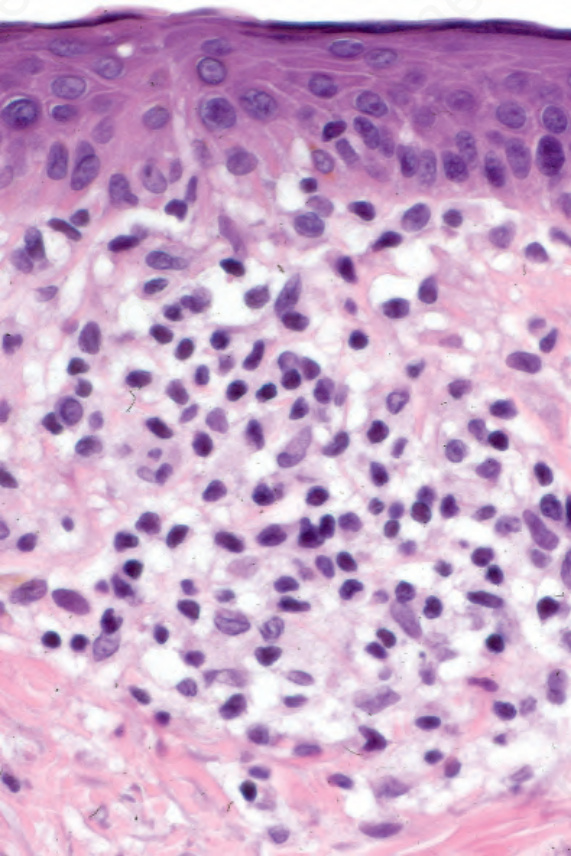
Fig. 29.25 Mycosis fungoides: mild cytological atypia is evident.
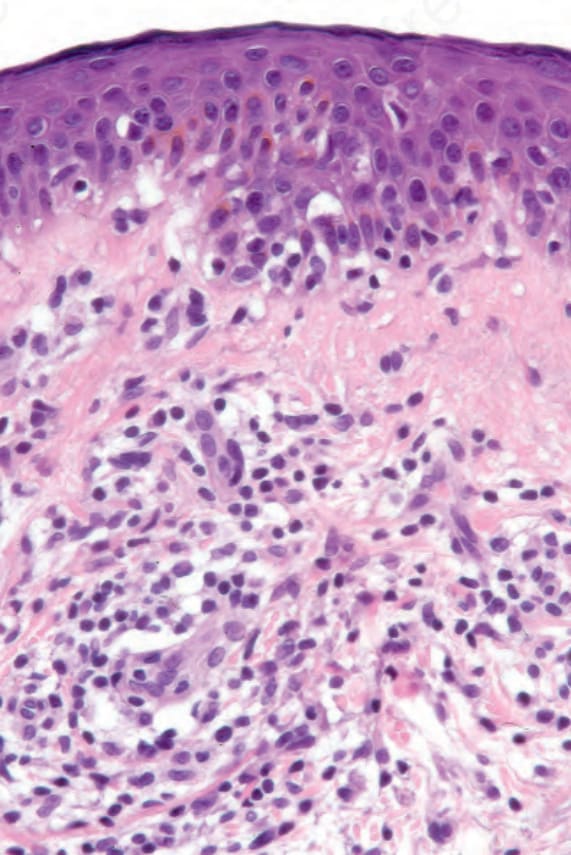
Fig. 29.26 Mycosis fungoides: there is just a hint of epidermotropism. This case illustrates the importance of clinicopathological correlation. A repeat biopsy following a period with no therapy showed typical features.
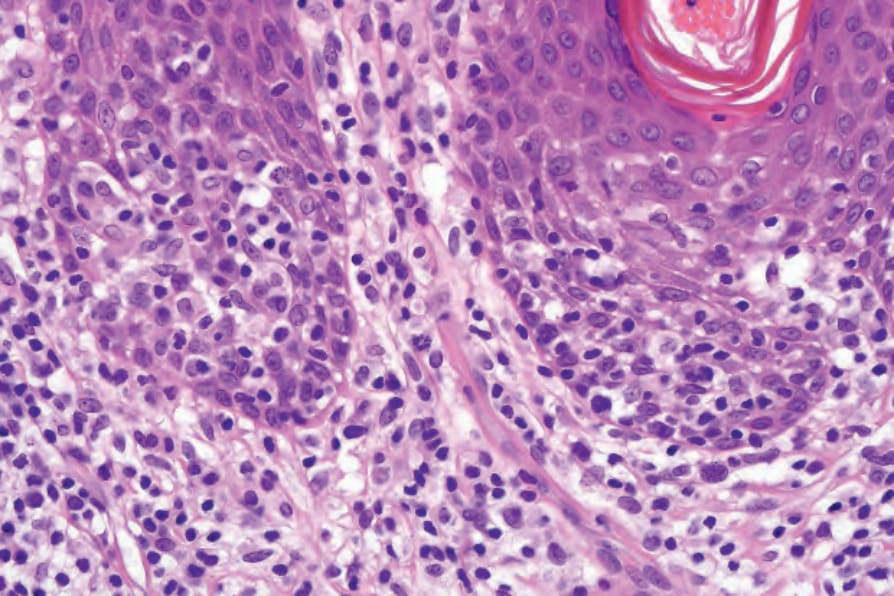
Fig. 29.29 Mycosis fungoides (plaque stage): there is a dense population of atypical lymphocytes with marked epidermotropism.
Fig. 29.33 Mycosis fungoides (plaque stage): in this example, the scarring is more marked.
Fig. 29.35 Mycosis fungoides (tumor stage): note the dissection of collagen.
Fig. 29.36 Mycosis fungoides (tumor stage): there is a uniform population of mycosis cells.
Fig. 29.37 Mycosis fungoides (tumor stage): this view demonstrates the conspicuous vasculature.
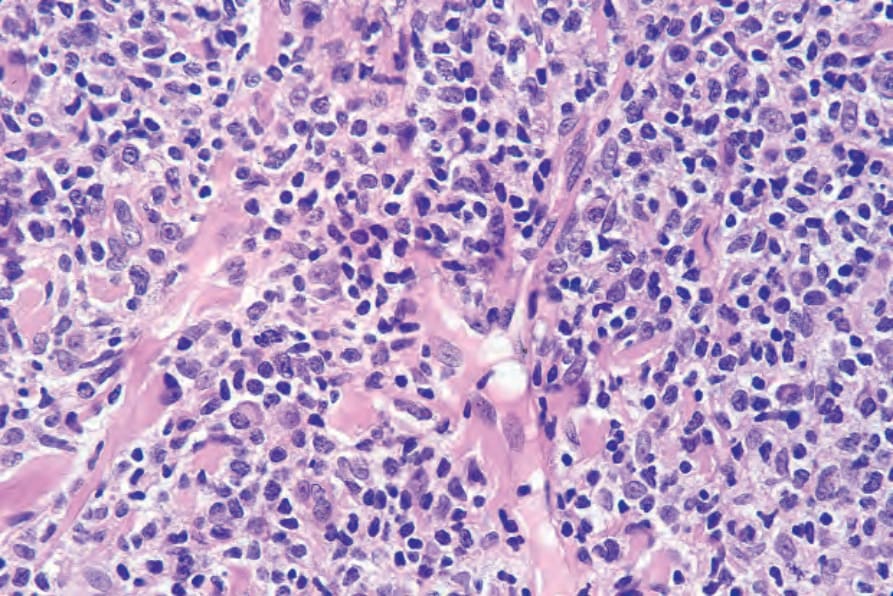
Fig. 29.38 Mycosis fungoides (tumor stage): high-power view.
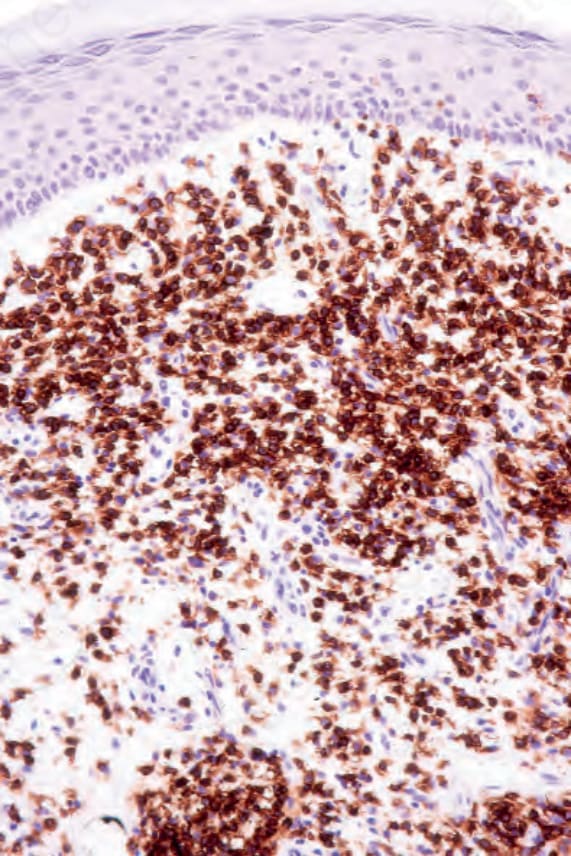
Fig. 29.40 Mycosis fungoides: the lymphocytes express CD4.
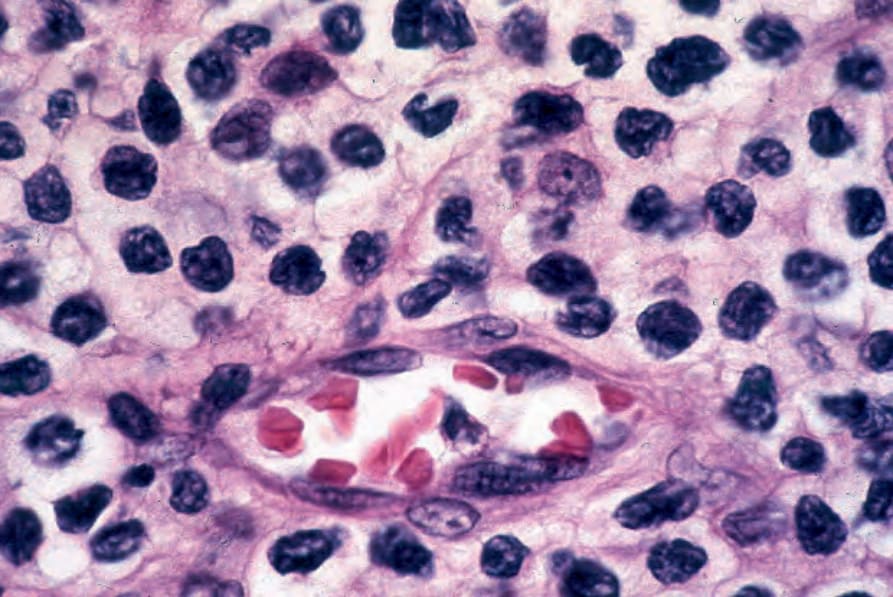
Fig. 29.44 Mycosis fungoides: mycosis cells surround a postcapillary venule.
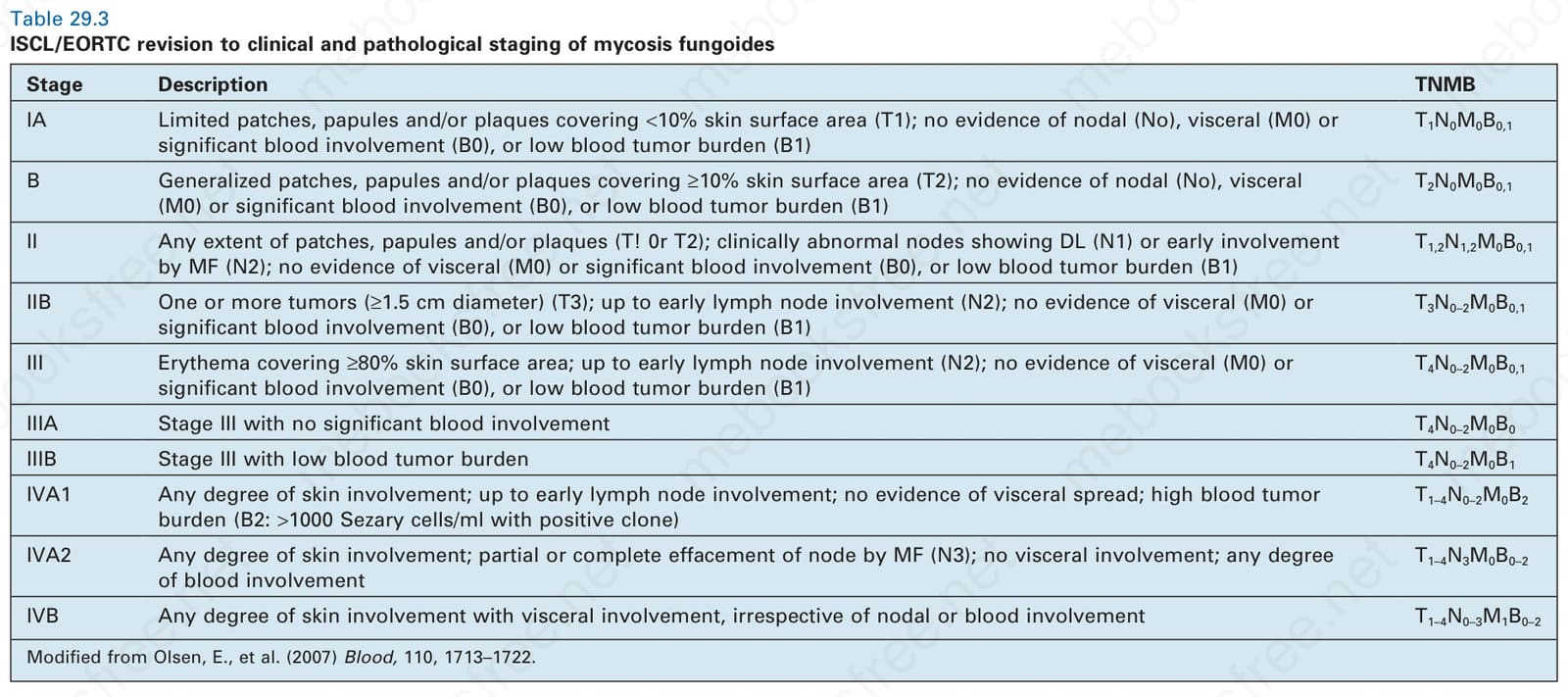
Table 29.3 ISCL/EORTC revision to clinical and pathological staging of mycosis fungoides
IVA1 Any degree of skin involvement; up to early lymph node involvement; no evidence of visceral spread; high blood tumor burden (B2: >1000 Sezary cells/ml with positive clone)
T1–4N0–2M0B2
IVA2 Any degree of skin involvement; partial or complete effacement of node by MF (N3); no visceral involvement; any degree of blood involvement
T1–4N3M0B0–2
IVB Any degree of skin involvement with visceral involvement, irrespective of nodal or blood involvement T1–4N0–3M1B0–2
Modified from Olsen, E., et al. (2007) Blood, 110, 1713–1722.
As with many malignancies, the etiology and pathogenesis of mycosis fungoides is likely to be multifactorial. Specific causative agents and precise pathogenetics are unknown.67
There is little evidence of genetic predisposition to mycosis fungoides. Familial occurrence is exceptional.68,69 However, a number of studies have inconsistently linked various human leukocyte antigen (HLA) types with mycosis fungoides. For example, HLA-B8 and HLA-AW19 have been found with increased frequency in mycosis fungoides patients from the United Kingdom, whereas an association with HLA-DR5 (DRB1*11) and
DQB1*03 was demonstrated in North American Caucasians.70–73 In studies from Israel, no association with HLA class I alleles was detected.74
It is thought that mycosis fungoides most probably develops as a consequence of chronic antigenic stimulation, either to long-term exposure to irritant substances or infective agents. Epidemiological studies have variously pointed toward occupational exposure to metals, plastics, cutting oils, solvents, glass, pottery, and ceramics, or working as a crop or vegetable farmer, painter, woodworker, or carpenter.10,75–79 A pathogenetic role for infective agents has also long been postulated.3,67,80 A wide range of potential agents,
1410 Cutaneous lymphoproliferative diseases and related disorders
particularly viruses, have been investigated including human T-cell lymphotropic virus-I (HTLV-I), HTLV-II, Epstein-Barr virus (EBV), cytomegalovirus (CMV), human herpes virus (HHV) 6, HHV7, and HHV8. However, the results of various studies looking for evidence of these agents in MF have produced conflicting or inconclusive results.6,67,81–84 Other organisms that have also been considered include S. aureus, persistent chlamydia infection, and Borrelia burgdorferi.76,85–88 However, there is no strong evidence in favor of these agents playing a direct role in MF pathogenesis.67 Nevertheless, it remains possible that various infectious agents are important initiators of the chronic lymphoproliferation from which lymphoma eventually develops, and a recent study has suggested that persistence of multiple infectious agents may be more important than any single one in isolation.89
A variety of recurrent genetic abnormalities have been identified in mycosis fungoides, although none are specific for this entity. They include deletions of chromosomes 1p, 17p, 10q, and 19, as well as gains on 4q, 18, and 17q.90,91 Alterations in a number of tumor suppressor and apoptosis-related genes have also been detected. Silencing of p15, p16, Nav3, PTEN, and p53 due to mutations, promoter hypermethylation, or allelic loss have all been implicated in the progression from plaque to tumor stage.92–96 More recently, massive parallel sequencing approaches have uncovered recurrent mutations in a multiple genes involved in epigenetic regulation, T-cell receptor signaling and particularly the JAK-STAT and NF-κB pathways.97–100 These alterations are reflected, to a certain extent, by the gene expression profile and phenotype of the tumor cells in mycosis fungoides which display features of at least partially activated skin resident effector memory cells, with a unique expression of chemokine receptors and adhesion molecules, and a bias toward a Th2-type phenotype. These exclusive properties help explain some of the characteristic features of MF, and provide insight into the evolution and progression of the disease.
The neoplastic cells express cytokine receptors CCR4 and CCR10.101,102 CCL17 and CCL22 are ligands for CCR4 and are present in high levels in skin lesions of affected patients.90,101 CCL17 is also increased in the serum of patients with the disease and Sézary syndrome, as is CCL27, the ligand for CCR10.67 Interaction of CCR4 and CCR10 with their respective ligands not only influences T-cell migratory properties, but promotes their survival via downstream activation of antiapoptotic pathways such as phosphatidylinositol-3-kinase and Akt.67 Such chemokine interactions are therefore likely to be important in initiation and perpetuation of the skin lesions and may explain the marked propensity for the malignant cells to localize in the epidermis.90
MF cells also express cell surface adhesion molecules such as CD45RO and IL-2R, and there is evidence of constitutive activation of several intracellular signaling proteins in the JAK-STAT family of molecules, similar to that witnessed following stimulation of normal T cells via the IL-2 receptor.91,103–107 This is likely to affect regulation of various aspects of cell survival and proliferation. Other factors may also contribute toward defects in normal apoptotic pathways in MF cells. For example, mutations of the Fas gene or abnormal splice variants of its transcript have been documented in MF and may protect against Fas-mediated stimulation of apoptosis.92,108 Also, gene expression profiling studies have generated a genetic signature specific for MF. This is characterized by multiple genes involved in the tumor necrosis factor (TNF) signaling pathway, including several inhibitors of apoptosis.109 Abnormalities in Fas and TNF signaling pathways may also contribute toward the constitutive activation of the NF-kappa B pathway noted in MF, further supporting a role for resistance to apoptosis in its pathogenesis.110
contribute to suppressing the host antitumor immune response and partly account for the increased risk of infections and second malignancies.115–118 Copy number gains and amplifications of JUNB, and JUNB overexpression have been documented in MF and may contribute to the Th2 bias, as JUNB promotes IL-4-mediated TH2 lymphocyte differentiation.119,120
The lymphocytes of MF are mature T cells that have transited through the lymph node and undergone antigenic stimulation. They show a particular tendency to colonize the epidermis (epidermotropism), although this is more evident in the patch and plaque forms than in tumor-stage disease where epidermotropism can be completely lost. To a lesser extent in early lesions, and more obviously in later stages, the infiltrate contains large cells with highly irregular, convoluted, or cerebriform nuclei, known as Sézary or mycosis cells (Fig. 29.15). Assessing their significance must be tempered with caution because identical cells (albeit in small numbers) are sometimes seen in the dermal infiltrate of a variety of dermatoses. Their presence must obviously be considered in the context of the accompanying histologic features and, in particular, in the light of the clinical information. Electron microscopically, mycosis/Sézary cells are characterized by a multilobed hyperconvoluted nucleus with conspicuous peripheral chromatin margination (Fig. 29.16).
Tumor cells in MF also seem equipped to evade the host immune system. Cytotoxic T cells can induce apoptosis via engagement of Fas, expressed on the surface of the target cell, by Fas ligand (FasL). The aforementioned defects in FasL signaling in MF cells are therefore likely to protect against tumor-specific cytotoxic T-cell-mediated immunity.90,92,108 Furthermore, the malignant cells in MF may express FasL, giving them the potential to eliminate tumor-specific CD8-positive cytotoxic T cells.111 They also show down-regulation of Th1-associated genes, and express cytokines such as IL-4, IL-5, and IL-18, most in keeping with Th2-type T cells.112–114 This Th2 bias, together with production of other immunomodulatory cytokines such as IL-10, transforming growth factor (TGF)-β, and soluble IL-2R may also
Epidermotropism is, therefore, the histologic hallmark of MF.121–130 It is related to the stage of the disease and the degree of differentiation of the
1411 Mycosis fungoides
lymphocytes. With the development of tumor stage MF, large transformed cells are conspicuous and epidermotropism is commonly lost. Epidermotropism as seen in cutaneous T-cell lymphoma differs from exocytosis of lymphocytes as seen in dermatitis/eczema in that there is usually no or only very mild spongiosis and vesiculation is not usually a feature. The presence of atypical lymphoid cells in an intraepidermal vesicle (the Pautrier microabscess) is typical, although not diagnostic, of mycosis fungoides (Fig. 29.17). Similar features may be seen, though less often, in Sézary syndrome, adult T-cell leukemia/lymphoma (ATLL), actinic reticuloid, and drug-induced T-cell pseudolymphomas.
29.19). Occasionally, small numbers of larger typical cells may also be evident. Palisading of lymphocytes along the basal layer of the epidermis is a diagnostic pointer. Individual necrotic keratinocytes are seen in some cases. A lymphohistiocytic infiltrate surrounds the vessels of the superficial vascular plexus and extends into the papillary dermis (Fig. 29.20). Eosinophils and plasma cells are present in small numbers or absent.128,131 Red cell extravasation is sometimes evident. Pigmentary incontinence is common. The connective tissue of the papillary dermis is occasionally increased with coarsening of the collagen bundles (although this is more typical of plaque stage disease).128 In the more advanced patch stage, increasing numbers of atypical cells may make the diagnosis more obvious, although frank Pautrier microabscesses are uncommon (Figs 29.21 and 29.22).
Patch stage mycosis fungoides The histopathological features of the early patch stage of MF are usually subtle and easily overlooked (Fig. 29.18). Multiple biopsies are commonly necessary to reach a diagnosis. The epidermis may be of normal thickness, slightly acanthotic, or less commonly atrophic, and often there is mild hyperkeratosis with focal parakeratosis. Basal cell hydropic degeneration is sometimes noted. Within the epidermis, there are characteristically small numbers of atypical irregular lymphoid cells, each surrounded by a clear halo, although in very early lesions they may sometimes be absent (Fig.
Sometimes, prior treatment with steroids or PUVA before biopsy masks the features, making the diagnosis virtually impossible, emphasizing the necessity for careful clinicopathological correlation (Figs 29.23–29.26).
Plaque stage mycosis fungoides In established plaque stage disease, there is usually little difficulty in establishing the correct diagnosis. There is compact hyperkeratosis, patchy parakeratosis, and the epidermis is commonly acanthotic, frequently adopting
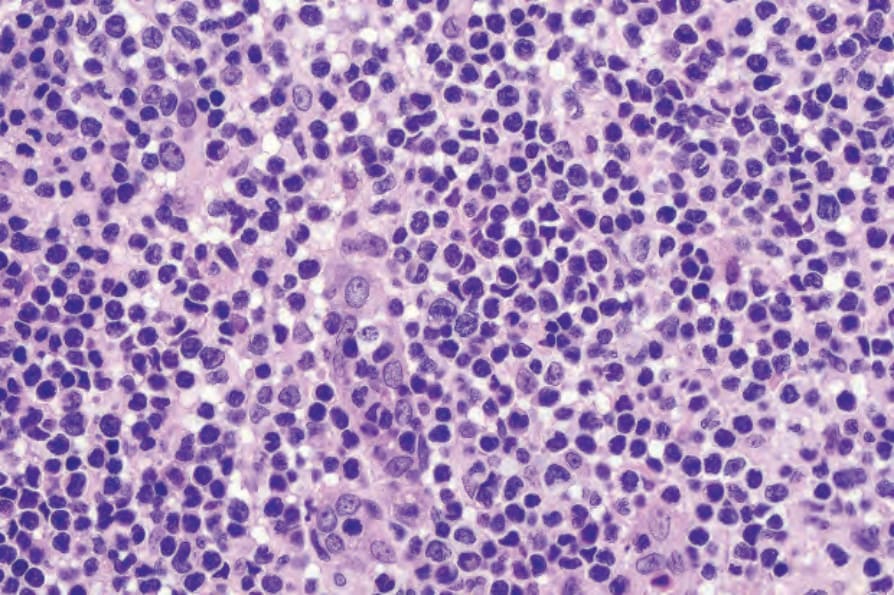
Fig. 29.15 Mycosis fungoides: the nuclei of the mycosis cells are hyperchromatic and highly irregular.
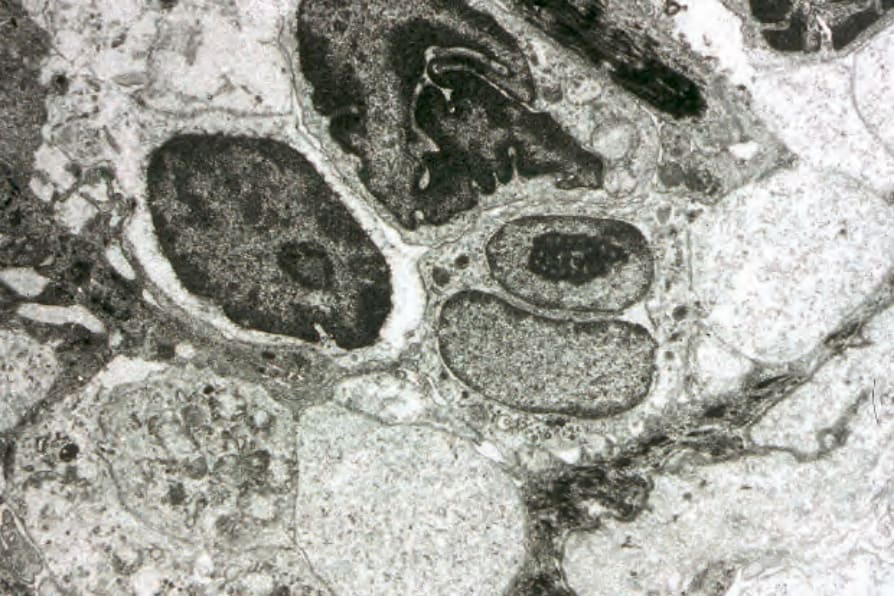
Fig. 29.16 Mycosis cells: note the highly convoluted (cerebriform nuclei).
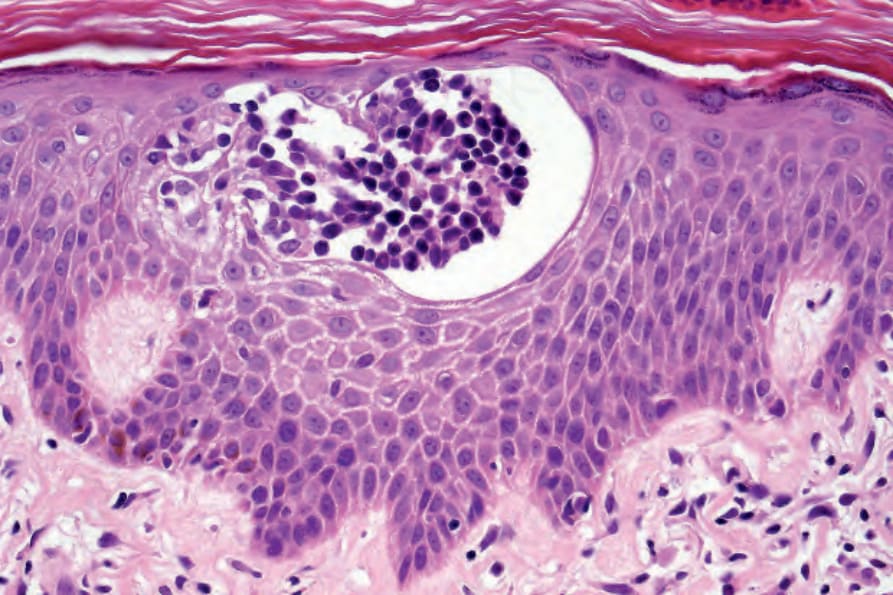
Fig. 29.17 Mycosis fungoides: a typical Pautrier microabscess composed of hyperchromatic atypical lymphocytes.
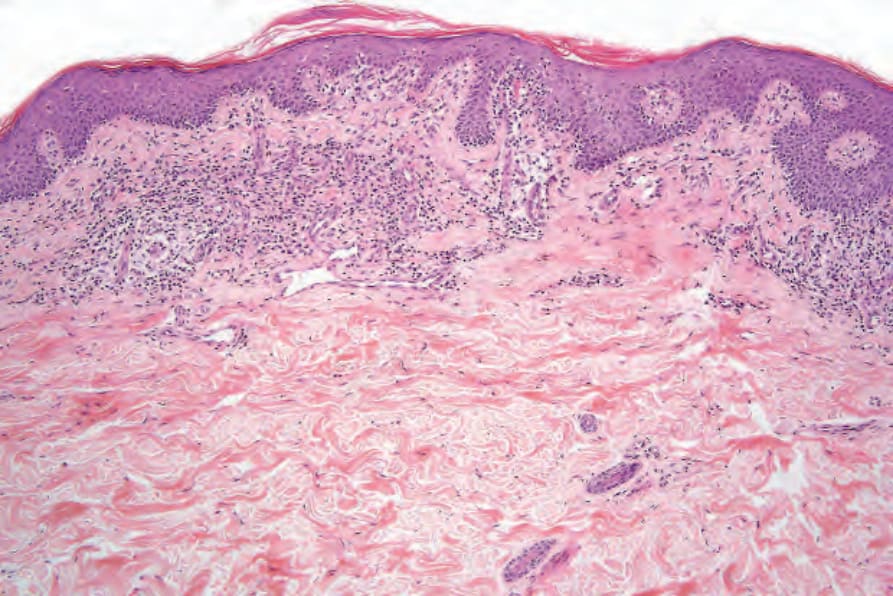
Fig. 29.18 Mycosis fungoides (patch stage): this biopsy of an early lesion shows focal parakeratosis, acanthosis, atypical lymphocytes ‘tagging’ the dermal–epidermal junction and a superficial perivascular lymphohistiocytic infiltrate.
Fig. 29.20 Mycosis fungoides (patch stage): there is a superficial perivascular lymphohistiocytic infiltrate. Atypical cells are not seen.
Fig. 29.21 Mycosis fungoides (patch stage): the epidermis shows psoriasiform hyperplasia in this more advanced example. Even at this magnification, epidermotropism is conspicuous.
Fig. 29.23 Mycosis fungoides: the case illustrated here and in Figs 29.24–29.26 demonstrates the effects of prior treatment with PUVA before biopsy. Note the dermal perivascular and interstitial lymphocytic infiltrate.
1412 Cutaneous lymphoproliferative diseases and related disorders
1413 Mycosis fungoides
In addition to the epidermal changes, follicular epithelium may be involved, and sometimes follicular mucinosis is evident. There may also be infiltration of sweat duct epithelium by the malignant cells (syringotropism).
In poikilodermatous lesions, the epidermis is typically flattened, atrophic, and covered by a scale of hyperkeratosis or parakeratosis (Fig. 29.30). Hypergranulosis is not usually a feature. Necrotic keratinocytes are occasionally seen. There is often hydropic degeneration of the basal layer of the epidermis (Fig. 29.31). Epidermotropism is a constant feature and occasionally mycosis/Sézary cells may be noted. In the dermis, there is a bandlike or perivascular lymphohistiocytic infiltrate. Pigmentary incontinence is usually a feature, and often telangiectatic vessels are evident. The superficial dermis may be scarred.
a psoriasiform appearance (Fig. 29.27). The infiltrate is much more intense than in the patch stage, and large numbers of atypical mononuclear cells are commonly present in the epidermis (Figs 29.28 and 29.29). Occasionally, however, epidermotropism is absent, particularly in patients who have been treated topically. In the dermis, the distribution is predominantly superficial and bandlike in character. Mycosis/Sézary cells may be present singly or in clusters or exceptionally replace almost the entire epidermis.
Pautrier microabscesses are not uncommon, being identified in 17–37.5% of cases.127 The infiltrate may obscure the dermal–epidermal junction, and cytoid bodies are sometimes present. Occasionally, the additional histologic features of confluent hyperkeratosis, irregular acanthosis, extensive basal cell hydropic degeneration, and apoptosis mimic lichen planus.
Verrucous lesions which show pseudoepitheliomatous hyperplasia and crusting with associated hyperkeratosis and parakeratosis may be clinically confused with deep mycoses and even keratoacanthoma.
The dermal infiltrate often contains an admixture of reactive inflammatory cells, including eosinophils, plasma cells, and histiocytes, in addition to atypical lymphocytes.127 Occasionally, multinucleate giant cells are evident and mitoses are sometimes seen. Coarse collagen bundles with or without increased numbers of fibroblasts are commonly present in the papillary dermis (Figs 29.32 and 29.33).
1414 Cutaneous lymphoproliferative diseases and related disorders
Tumor stage mycosis fungoides In tumor stage MF, a very dense infiltrate occupies the dermis, sometimes extending into the subcutaneous fat (Figs 29.34–29.38). A top-heavy configuration may be apparent. The lesions are often ulcerated, and epidermotropism is typically either slight or absent. The infiltrate, which may be diffuse or show an ill-defined nodular outline, in addition to containing mycosis/ Sézary cells, often has large numbers of highly pleomorphic cells. Mitotic figures are conspicuous and frequently abnormal.
A broad spectrum of histologic changes has been described in biopsies of early stage MF, and many of these overlap with features seen in inflammatory dermatoses.131,132 The following have been reported to be useful when trying to establish a definite diagnosis in such situations:123,124,127,133
• lymphocytes in linear array at the dermal–epidermal junction (linear epidermotropism),
• hyperconvoluted lymphocytes within the epidermis (epidermal cerebriform cells),
• disproportionate epidermotropism,
• Pautrier microabscesses,
• epidermal lymphocytes larger than dermal lymphocytes,
• wiry bundles of papillary dermal collagen associated with a lichenoid infiltrate,
• lymphocyte atypia, although characteristic, is often not apparent in early lesions,
• mitotic activity is of little value in early disease. However, two multivariate analyzes produced conflicting results as to which of these features was the most helpful.127,133 This emphasizes the need for good clinicopathological correlation, and is the reason why diagnostic algorithms incorporating clinical, pathological, and molecular findings have been proposed (see below). It is also important to be aware of a patient’s current treatment when assessing a biopsy specimen, since local treatments may mask any tendency to epidermotropism.
1415 Mycosis fungoides
MF is commonly characterized by accumulation and proliferation of CD4+, CD45RO-positive helper/memory T lymphocytes, although occasionally a CD8+ and even CD4/CD8-phenotypes may be seen (Figs 29.39 and 29.40). The latter has no bearing on prognosis. The lymphocytes usually also express the pan-T-cell antigens CD2, CD3, CD5, and CD7, as well as TCRαβ and cutaneous lymphocyte antigen (CLA).96,111 CD7 is often focally lost very early, and it is not of diagnostic value as its expression is also often lost in reactive conditions (Fig. 29.41), as are less frequently CD2, CD3, or CD5.24,134 CD25 (IL-2 receptor) is positive in up to 50% of cases, and the T-follicular helper cell marker, PD1, is also frequently expressed.135,136 CD30 expression correlates with transformation but has no prognostic implications. Unlike some other cutaneous T-cell lymphomas, MUM1, as identified by the MUM1p antibody, is rarely expressed.137 Aberrant expression of CD20 is occasionally seen, and this is not associated with expression of
1416 Cutaneous lymphoproliferative diseases and related disorders
other B-cell markers.138 Expression of TCRγδ is rarely found, and this has no bearing on behavior or prognosis in the context of classic cutaneous T-cell lymphoma (mycosis fungoides).139
Evidence of a clonal T-cell receptor gene rearrangement may be identified by Southern blot or polymerase chain reaction (PCR) in the majority of cases.53–60 Overall, TCR gene rearrangements can be anticipated in up to 100% of cases of tumor stage, 50–100% of plaque stage, and 50–78% of patch stage MF.43 The results should, however, be interpreted with caution since TCR gene rearrangements have been described in a number of inflammatory dermatoses including discoid lupus erythematosus, lichen planus, lichen sclerosus, and pityriasis lichenoides et varioliformis acuta (PLEVA).58–64
Dermatopathic lymphadenopathy with lymph node involvement Lymphadenopathy is frequently present in MF but does not always correlate with histologic evidence of lymphomatous involvement. The features of dermatopathic lymphadenitis are characterized by a marked infiltration of the nodal paracortical region by large numbers of histiocytes, including interdigitating reticulum cells and Langerhans cells.140–144 Occasional plasma cells and eosinophils may be present. The histiocytes often contain melanin. At this stage, there is no distortion of the lymph node architecture. With progression, careful study of the paracortex may reveal infiltration by mycosis/ Sézary cells (Fig. 29.42). These are best identified around the postcapillary venules (Figs 29.43 and 29.44). At first, they may be present singly or in
1417 Transformation of mycosis fungoides
Updated ISCL/EORTC classification Dutch system NCI-VA classification
N1 Grade 1: dermatopathic lymphadenopathy
LN0: no atypical lymphocytes seen LN1: scattered atypical lymphocytes, not in clusters LN2: many atypical lymphocytes arranged in clusters of 3–6 cells
N2 Grade 2: early involvement by mycosis fungoides
LN3: aggregates of atypical lymphocytes observed but lymph node architecture preserved
N3 Grade 3: lymph node architecture partially effaced by lymphoma Grade 4: complete effacement of lymph node architecture by lymphoma
LN4: partial or complete effacement of the lymph node architecture by lymphoma
Modified from Olsen, E., et al. (2007) Blood, 110, 1713–1722.
• N1: clinically abnormal nodes; dermatopathic lymphadenopathy; atypically lymphocytes may be present but only singly or in small clusters (3–6 cells), – N1aNo: evidence of monoclonal T-cell population by PCR, – N1b: clonal T-cell population detected by PCR,
• N2: clinically abnormal nodes; aggregates of atypical cells present but nodal architecture preserved, – N2aNo: evidence of monoclonal T-cell population by PCR, – N2b: clonal T-cell population detected by PCR,
• N3: clinically abnormal nodes; partial or complete effacement of nodal architecture,
• Nx: clinically abnormal nodes; no histologic confirmation.
small groups, but in advanced disease sheets of tumor cells are evident and the nodal architecture is effaced.
Historically, two histologic staging systems have been used to document the degree of lymph node involvement. Both record the number and distribution of ‘abnormal’ lymphocytes within the nodal parenchyma, but differ in their definition of what constitutes ‘abnormal’. In the NVI/VA system, neoplastic cells are defined as lymphocytes with cerebriform, irregularly folded, hyperconvoluted nuclei irrespective of size.145,146 The Dutch system only accepts cerebriform cells >7.5 microns in diameter as neoplastic.144 Both of these approaches have been incorporated into a simplified version in the proposed ISCL/EORTC staging system.30 In this, minor degrees of lymph node involvement are grouped with changes of dermatopathic lymphadenopathy, since the prognostic impact of the former is unclear.30 Partial and complete effacement of lymph node architecture clearly impacts on survival and is worthy of separate consideration.30 The results of clonality studies can also be included in lymph nodes showing possible early involvement. However, there is no clear evidence that the presence of a clone detected by PCR in an uninvolved or minimally involved lymph node has any added impact on outcome. The definitions of the various stages are summarized below, and a comparison of the various systems is shown in Table 29.4:
• N0N: clinically abnormal nodes therefore no biopsy required (abnormal node ≥1.5 cm),
Fig. 29.27 Mycosis fungoides (plaque stage): there is focal parakeratosis and marked psoriasiform hyperplasia.
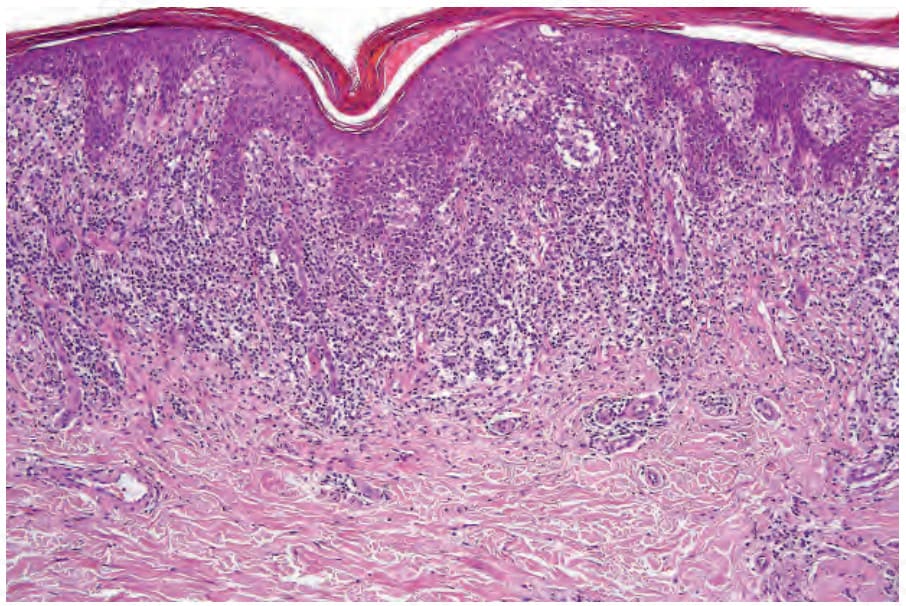
Fig. 29.28 Mycosis fungoides (plaque stage): there is a dense upper dermal bandlike infiltrate.
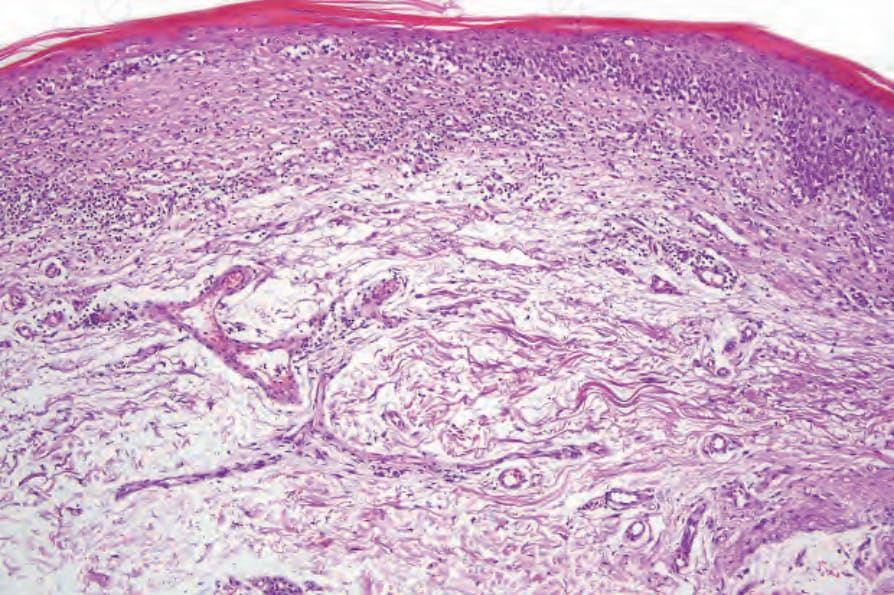
Fig. 29.30 Mycosis fungoides (poikiloderma atrophicans vasculare): there is hyperkeratosis, epidermal atrophy, marked basal cell hydropic degeneration, and fibrosis.
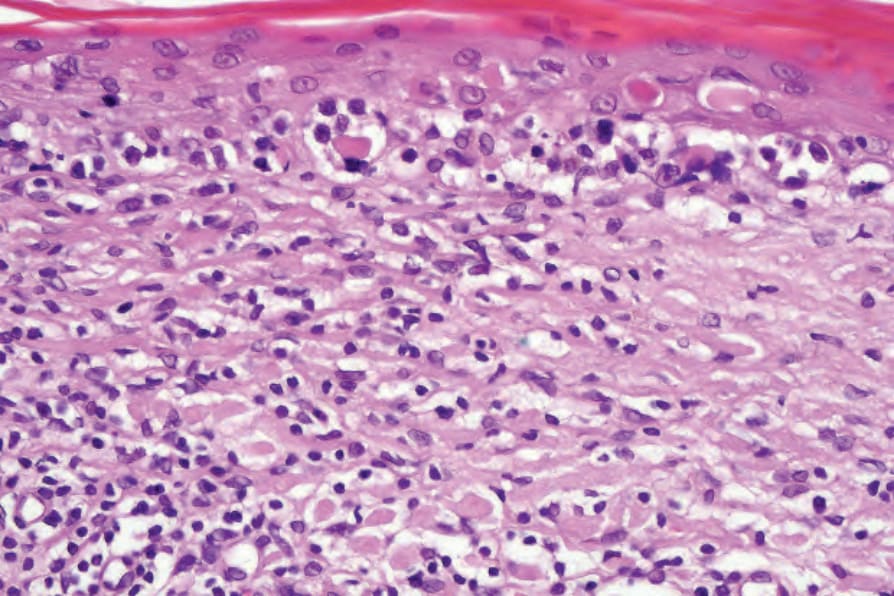
Fig. 29.31 Mycosis fungoides (poikiloderma atrophicans vasculare): there is liquefactive degeneration of the basal layer, and cytoid bodies are present. Note the scattered intraepidermal atypical lymphocytes.
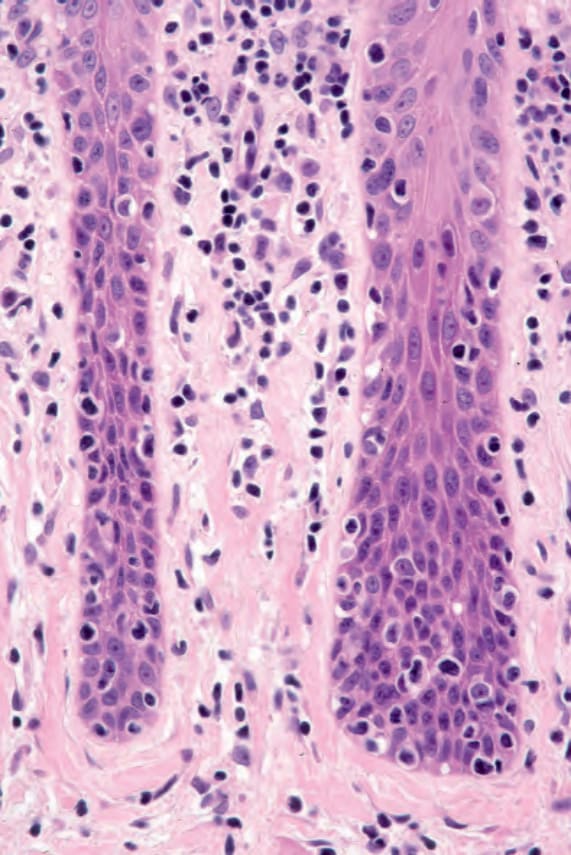
Fig. 29.32 Mycosis fungoides (plaque stage): note the vertically orientated fibrous tissue. This results from chronic scratching.
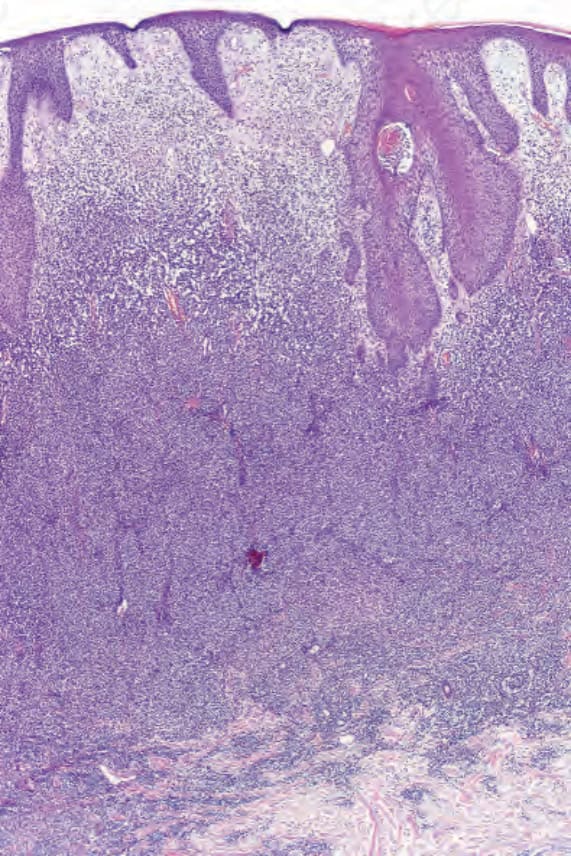
Fig. 29.34 Mycosis fungoides (tumor stage): an intense infiltrate is present in the dermis.
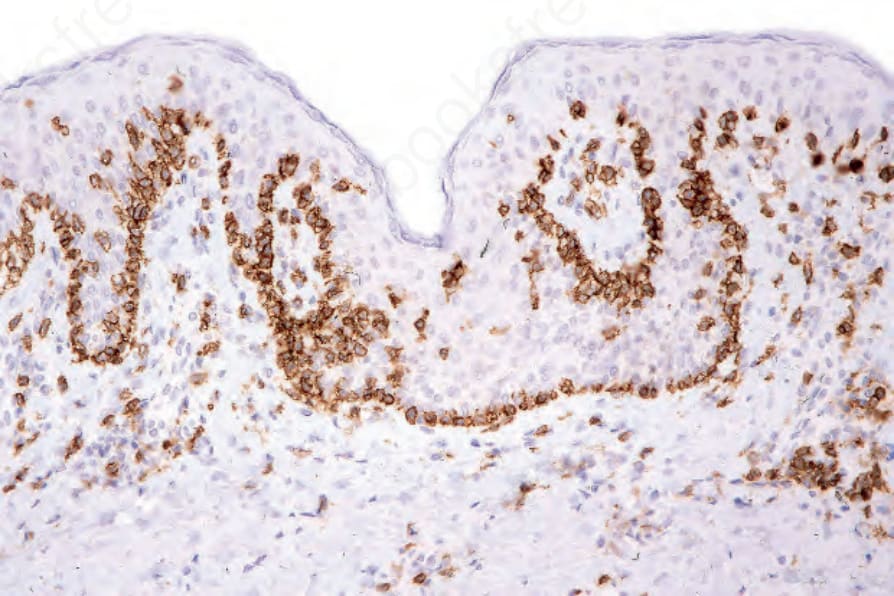
Fig. 29.39 Mycosis fungoides: intraepidermal lymphocytes are highlighted with CD3 immunohistochemistry. Note the conspicuous tagging.
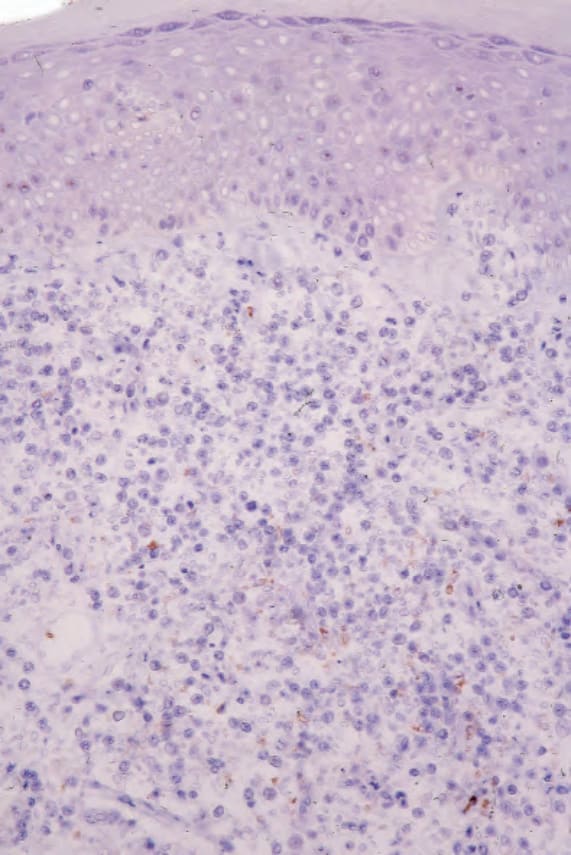
Fig. 29.41 Mycosis fungoides: CD7 is absent.
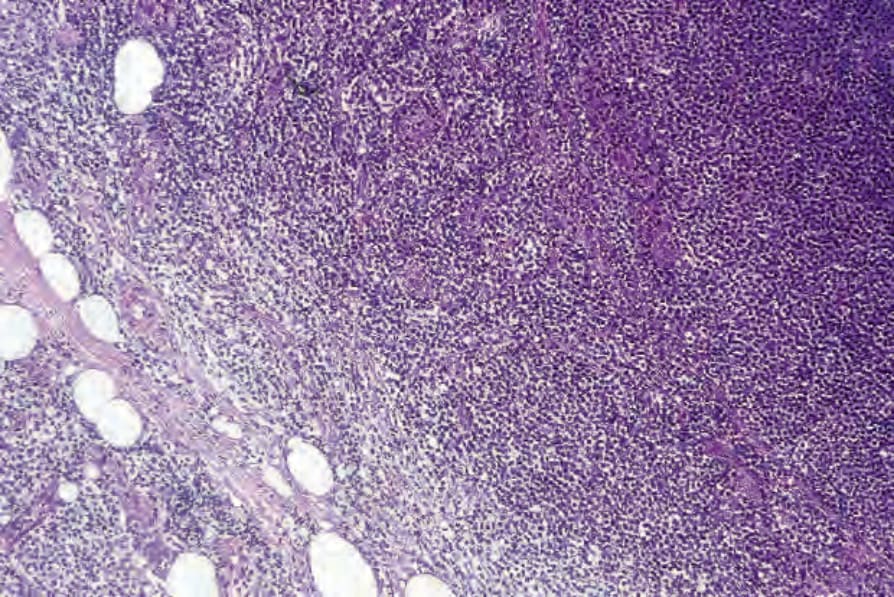
Fig. 29.42 Mycosis fungoides: this lymph node biopsy shows partial replacement by mycosis cells. Note the spillover into the pericapsular fat.
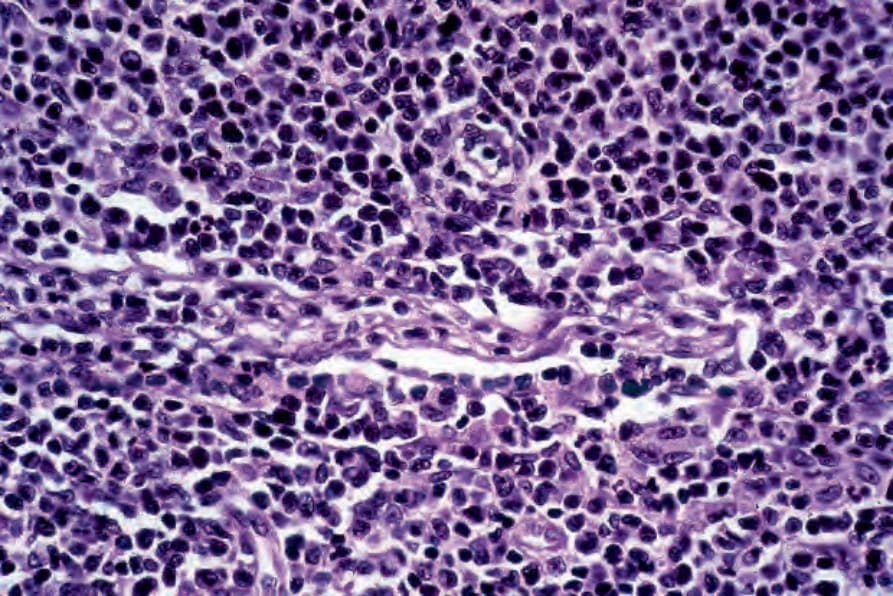
Fig. 29.43 Mycosis fungoides: higher power view shows a monomorphic infiltrate of Sézary cells.

Table 29.4 Pathological staging systems for lymph nodes in mycosis fungoides
Differential diagnosis Skin biopsies from patients with MF often show features that are also seen in various benign dermatoses.131,132 Patch stage mycosis fungoides must be distinguished in particular from chronic superficial dermatitis, which is typically a mild spongiotic process. Lymphocyte atypia and epidermotropism are not features. Lymphomatoid drug reactions may be histologically indistinguishable from MF. Differential diagnosis requires consideration of a constellation of clinical, histologic, immunohistochemical, and molecular features. This is recognized in the diagnostic algorithm recently proposed by the ISCL.24 This system assigns weighted scores to multiple criteria, up to a maximum of six, a diagnosis of MF requiring a total of at least four. This is an eminently sensible approach, but as yet there are only limited reports of its usefulness.
MF showing marked epidermotropism should be distinguished from CD8+ primary cutaneous epidermotropic T-cell lymphoma. ATLL commonly presents with histologic features indistinguishable from mycosis fungoides.
Tumor stage MF may be confused with other lymphomatous infiltrates, particularly if the cells are very pleomorphic and especially if epidermotropism is absent. Often, only careful review of the clinical information, taken in conjunction with the histologic features of previous biopsies (if available) and immunohistochemistry, allows the correct diagnosis to be made.