臨床特徵
- 蒙古斑(Mongolian spot)表現為相對均勻的青灰色(slate blue)不褪色變色區,邊緣波浪狀、形狀不規則,最常位於薦部。
- 較少見時分布更廣,如後大腿、小腿、背、肩。
- 通常出生時或不久後出現,最常見於日本人、中國人與深色人種;男性略多。
- 病灶可頗大,直徑達 10 cm;偶可疊加於另一蒙古斑之上。
相關共病
- 常伴多種共病,包括遺傳性代謝疾病、血管性胎記與隱性脊柱閉合不全(occult spinal dysraphism)。
- 最常見的潛在儲積病為 Hurler syndrome(mucopolysaccharidosis type II)、GM1 type I gangliosidosis、mucolipidosis type II,其次為 Niemann-Pick disease 與 mannosidosis。
- 蒙古斑合併血管性胎記(如 nevus flammeus)一般稱 phakomatosis pigmentovascularis。
- 亦曾與 noninvoluting congenital hemangioma、Sturge-Weber syndrome、Klippel-Trenaunay syndrome、cutis marmorata telangiectatica congenita、Sjögren-Larsson syndrome 及節段性 café-au-lait 斑併存報告。
組織病理特徵
- 蒙古斑與其他原發性真皮黑色素細胞病灶(dermal melanocytoses)被認為代表黑色素細胞自神經嵴向表皮的跨真皮遷移受阻。
- 病灶特徵為稀疏的真皮內樹突狀、色素多變的黑色素細胞,傾向平行於皮膚表面排列,主要位於深網狀真皮。
- 覆蓋的上皮正常。
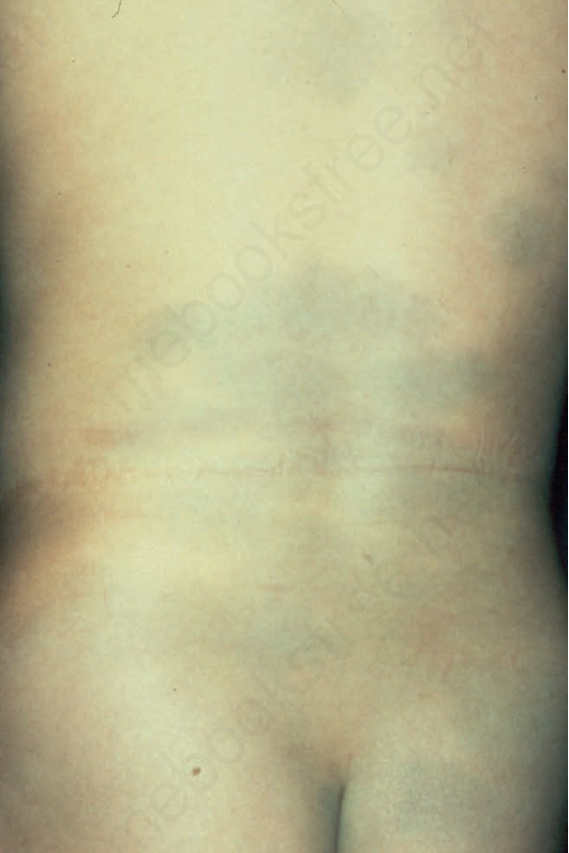
圖 25-217:蒙古藍斑,此孩童軀幹與臀部廣泛淡藍色變色。
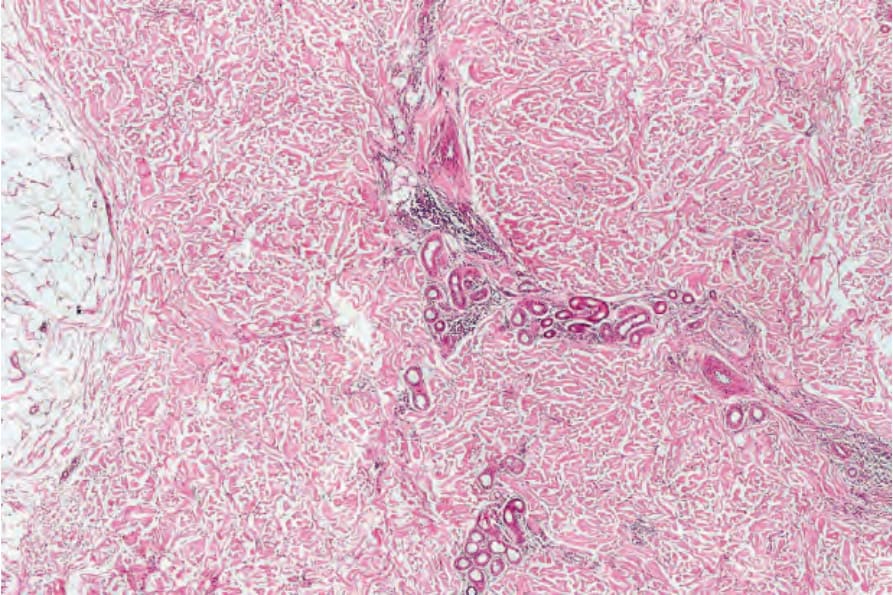
圖 25-218:蒙古藍斑,低倍檢查特徵細微,為深層真皮細胞密度增加。
病程與預後
- 多數蒙古斑於嬰兒期或孩童期自發退行,一般在青春期消失。
- 持續至成年罕見,通常與薦部外位置相關。
- 伴遺傳性儲積病者通常不退行,並可隨時間色素加深。
- 為良性樹突細胞增生,預後一般由共病決定。