定義與概念爭議
- 角化棘皮瘤(keratoacanthoma,molluscum sebaceum)為快速生長的皮膚腫瘤,主要發生於身體暴露面。
- 一般認為源於毛囊漏斗部 (follicular infundibular),嚴格說非表皮腫瘤,但因組織學與鱗狀細胞癌相似而納入本節。
- 命名與性質爭議甚多:曾被描述為良性腫瘤、偽惡性、退化性惡性,以及鱗狀細胞癌變異型。有案例記載持續性、復發、神經周圍浸潤、血管侵犯及轉移擴散。
- 雖多數視為鱗狀細胞癌變異型,此觀點仍具爭議、未被普遍接受。
臨床特徵與亞型 (Clinical features)
- 最常見為孤立型角化棘皮瘤 (solitary keratoacanthoma),主要見於白皮膚人種,亞洲人與非裔加勒比人罕受影響。
- 通常與過度 UVB 暴露相關,好發於日光受損皮膚,尤其臉部、前臂、腕及手背。
- 性別差異:男性多於女性 (2–3 : 1),發生率隨年齡增加,多數病人為第六或第七個十年;兒童罕見。典型鱗狀細胞癌發生率至少為角化棘皮瘤的三倍。
- 孤立型表現為平滑半球形丘疹,數週內迅速增大為 1- 至 2-cm 直徑、離散、圓或卵圓、常為肉色的臍狀結節,中央有角質充填火山口。色素沉著極罕。直徑大於 3 cm 者有時稱巨大角化棘皮瘤 (giant keratoacanthoma)。
- 通常隨後消退,腫瘤吸收、角質栓脫落,留下凹陷、色素減退疤痕。典型角化棘皮瘤壽命為 4–6 個月。手術介入後復發可見於高達 8% 的病人。
- 其他亞型:
- Agglomerate keratoacanthoma:數個病灶融合成單一大斑塊,可持續達 6 個月後退化。
- Keratoacanthoma centrifugum marginatum(多結節型):極罕見,以明顯周邊擴張為特徵,病灶可達直徑 20 cm;中央隨腫瘤擴張而癒合,消退常較久(6–12 個月)。部分宜視為低惡性度鱗狀細胞癌。
- Subungual keratoacanthoma(甲下型):源於甲基質而非毛囊漏斗部,行為較破壞性,多侵犯拇指與食指,呈疼痛、腫脹、紅斑,可似慢性甲溝炎;放射學示特徵性杯狀溶骨無骨膜反應;curettage 反應良好,復發率僅 14%。與 incontinentia pigmenti 的甲下腫瘤相同。
- 大量角化棘皮瘤的罕見症候群:
- Fergusson-Smith syndrome(家族性原發自癒性皮膚鱗狀上皮瘤):autosomal dominant,男性為女性三倍,常於兒童或成年早期發多發復發腫瘤。
- Grzybowski syndrome(疹發型):發大量(數百至數千)1–5 mm 毛囊性丘疹,性別發生率相等,常瘙癢有時疼痛,ectropion 為苦惱併發症。長期追蹤未見轉移潛能。
- Witten and Zak syndrome(混合型):合併上述兩型特徵。
- 疹發型病灶亦可發生於免疫抑制者;疹發性角化棘皮瘤與 pembrolizumab、laser resurfacing、植皮、光動力治療、leflunomide、刺青、ruxolitinib、quizartinib、imiquimod 及 sorafenib 有關,但這些與 Grzybowski 型無關。
- 孤立型角化棘皮瘤無證據顯示與內臟惡性腫瘤增加相關。
- 角化棘皮瘤可罕見發生於既存皮脂腺痣內或為 Muir-Torre syndrome 的表現。
致病機轉 (Pathogenesis)
- 可於多種實驗動物以煤焦油衍生物塗抹皮膚誘發;人類對應者見於長期接觸瀝青與焦油者,近期偶見於接受煤焦油製劑治療乾癬者。
- 為克隆性 (clonal),已辨識多種染色體異常,含 trisomy 7,1p、8q、9q 之增益,3p、9p、19p、19q 之缺失,及 2、8 號染色體易位;已注意到 RAS 突變 (HRAS 與 NRAS),部分病灶有微衛星不穩定。
- 多數腫瘤似為 UVB 媒介,多發生於日曬皮膚且常見光化損傷徵象。xeroderma pigmentosum 及慢性免疫抑制者(尤其腎移植後)發生率升高。
- 多數病灶 HPV DNA 陰性,少數可測得(types 5, 9, 10, 14, 16, 19, 20, 21, 25, 37, 38, 49, 80 等);免疫抑制者較免疫健全者更常測得 HPV。
組織學特徵 (Histologic features)
- 準確診斷完全仰賴足夠的臨床病史;缺乏病史或僅有組織碎片時極不宜下此診斷。
- 組織診斷仰賴辨識典型火山口狀 (crateriform) 結構,須行貫穿病灶中心的切除或深部切開活檢。成熟腫瘤對稱,具外生與內生成分,由常為大型的中央角質栓(有時與擴張毛囊漏斗部連續)伴顯著鱗狀上皮增生組成,病灶兩側表皮隆起成良好的衣領狀 (collarette)。
- 典型者以高分化、常淡染、嗜伊紅、玻璃樣細胞質、顯著傾向角化為特徵;細胞質內肝醣常豐富。可見壞死,常有含嗜中性球的微膿瘍。被陷入的彈性纖維(尤伴明顯日光彈性變性時)據稱具特徵性。
- 有絲分裂雖可明顯,但幾乎一律正常,主要見於腫瘤小葉周邊的增生上皮。生長極少超過汗腺深度,常伴血管性間質,常被淋巴球、組織球、漿細胞、嗜中性球及不等數目嗜伊紅球浸潤。
- 隨病灶老化與退化,角質栓喪失,增生上皮趨於變平,留下略呈乳頭狀的基底伴底層慢性發炎與纖維化;浸潤有時呈苔癬樣分布,主要由可能參與病灶退化的細胞毒性 T 細胞組成。退化常伴對釋出角質的異物巨細胞反應。退化機轉不確定,凋亡 (apoptosis) 可能特別重要。
- keratin 與 filaggrin 表現分析指向外毛根鞘 (outer root sheath) 分化。
- 罕由典型角化棘皮瘤產生經典鱗狀細胞癌,見於老年人。
鑑別診斷 (Differential diagnosis)
- 與鱗狀細胞癌有顯著相似,偶在臨床、甚至組織學上都無法區別。
- 流式細胞術增殖指數與 S-phase fractions 無法區別兩者;基質金屬蛋白酶、E-cadherin、catenins、syndecan-1 等亦僅見細微差異。VCAM 與 ICAM 表現可能有些區別價值。bcl-xL、TP53、COX-2 表現及端粒酶活性等資料顯示有顯著差異。此等皆僅為相對指引,區別需審慎觀察與臨床對照。
- 通常臨床病史與組織病理可作診斷並預測無虞結局;若對生物行為有任何疑慮,建議如鱗狀細胞癌般完整手術切除。
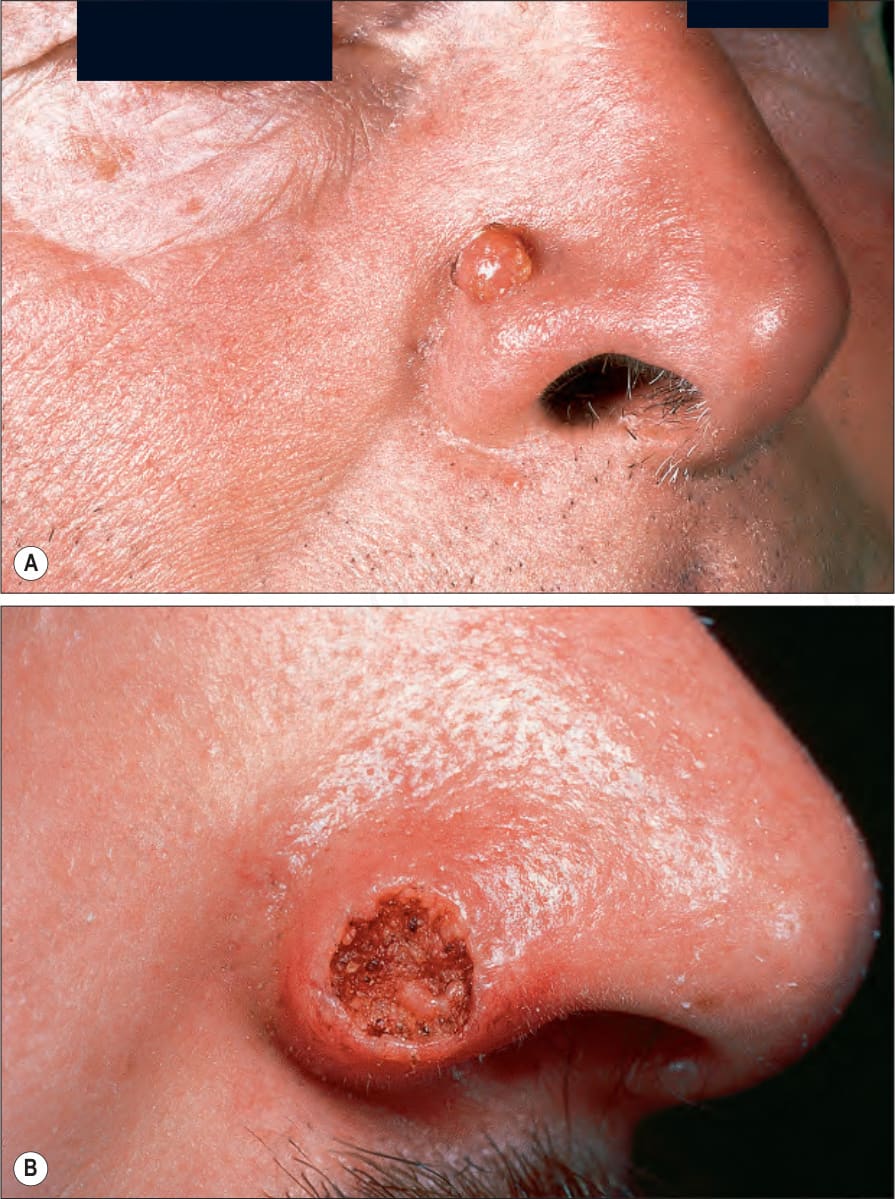
圖 24-177:角化棘皮瘤:(A) 鼻部典型圓頂狀病灶(常受侵犯部位);(B) 此例中央火山口特別發達。
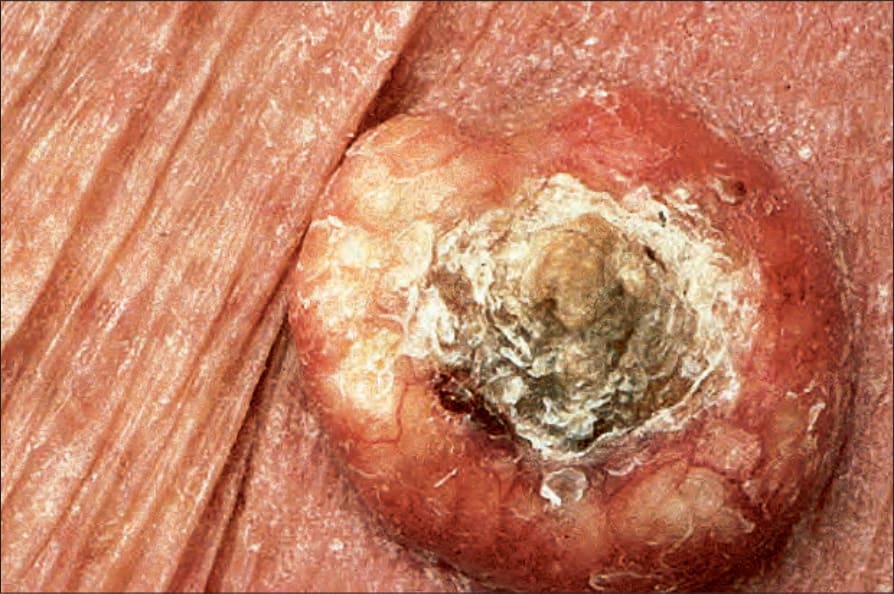
圖 24-178:角化棘皮瘤,角質栓近觀。
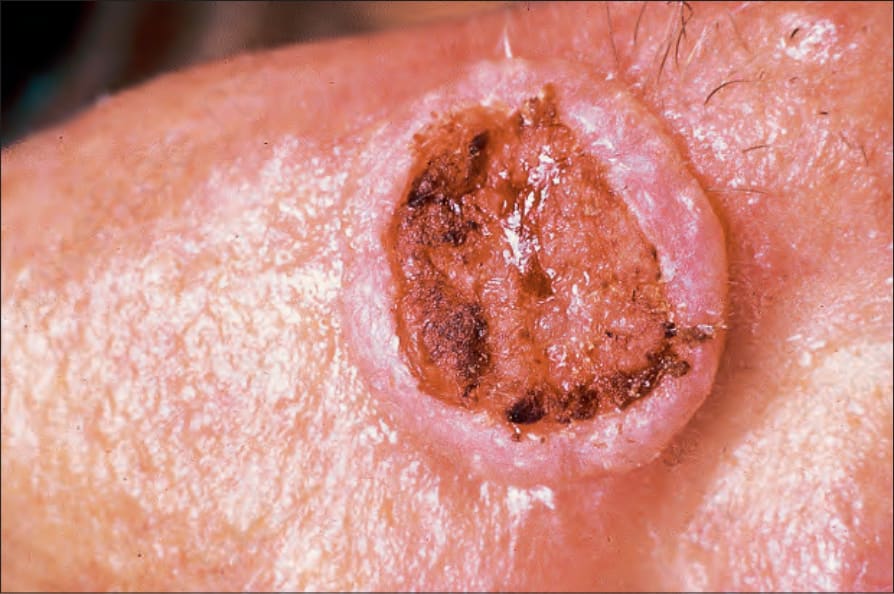
圖 24-179:角化棘皮瘤,此例可清楚見火山口邊緣。
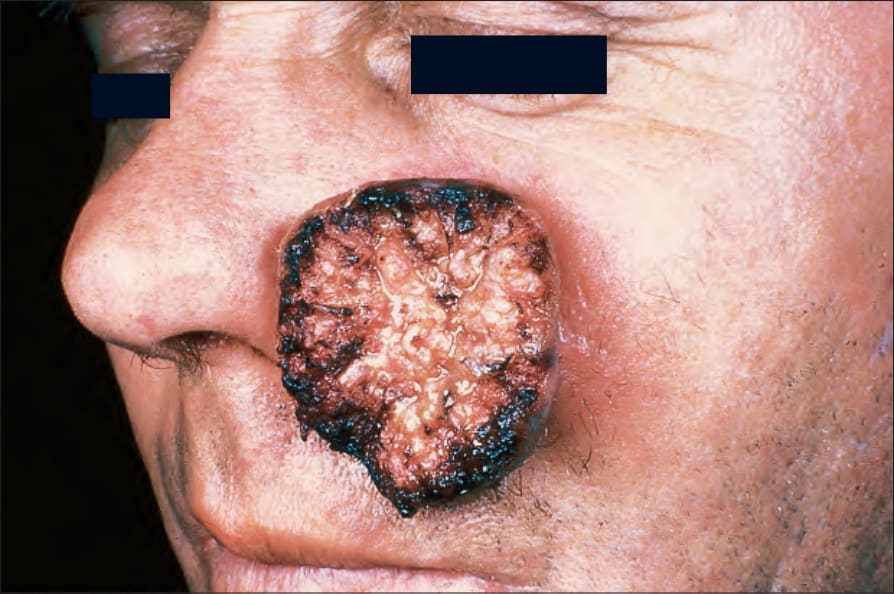
圖 24-180:巨大角化棘皮瘤,此類巨大變異型伴相當的組織損傷與疤痕。
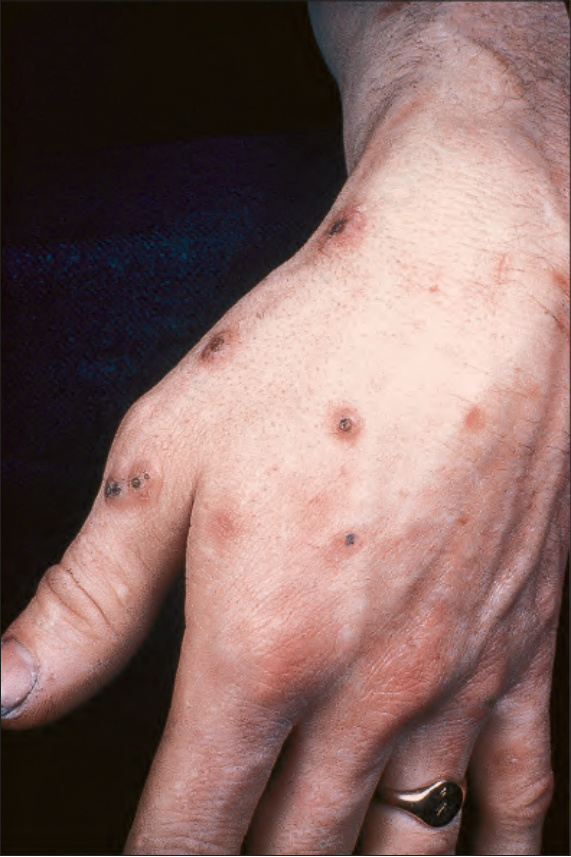
圖 24-181:Fergusson-Smith syndrome,可見眾多小型但典型的病灶。
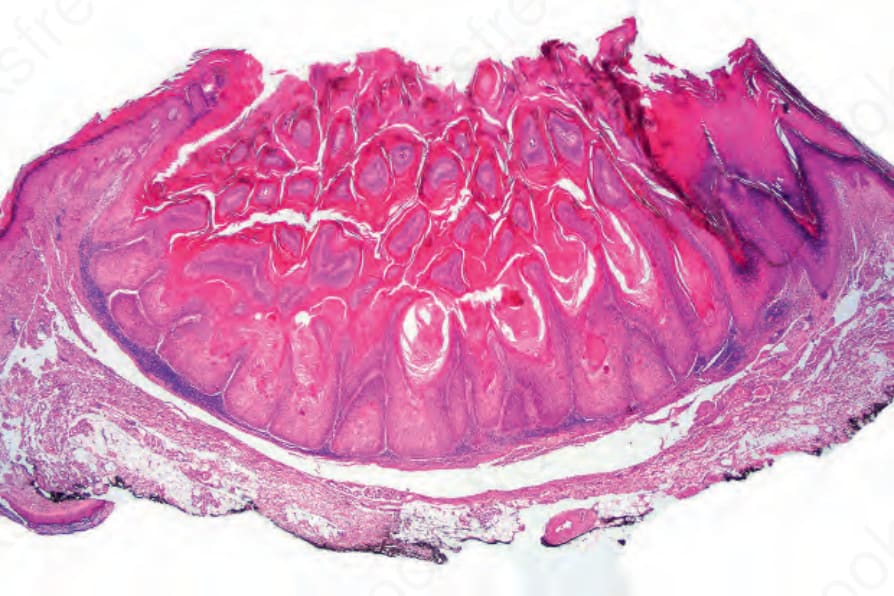
圖 24-182:角化棘皮瘤,典型者的掃描視野,顯示發達的衣領狀結構。
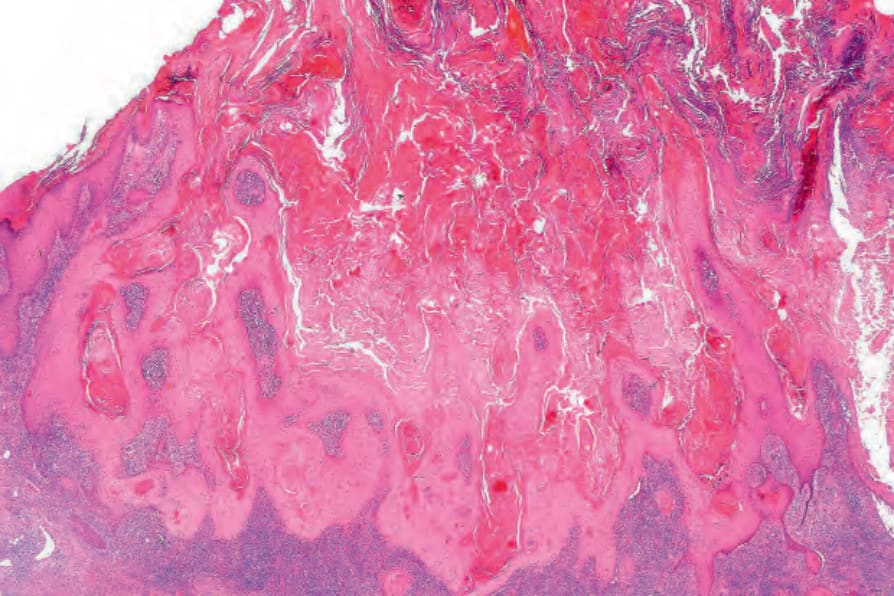
圖 24-183:角化棘皮瘤,此例顯示側方衣領、角質栓及底層增生的高分化鱗狀上皮。
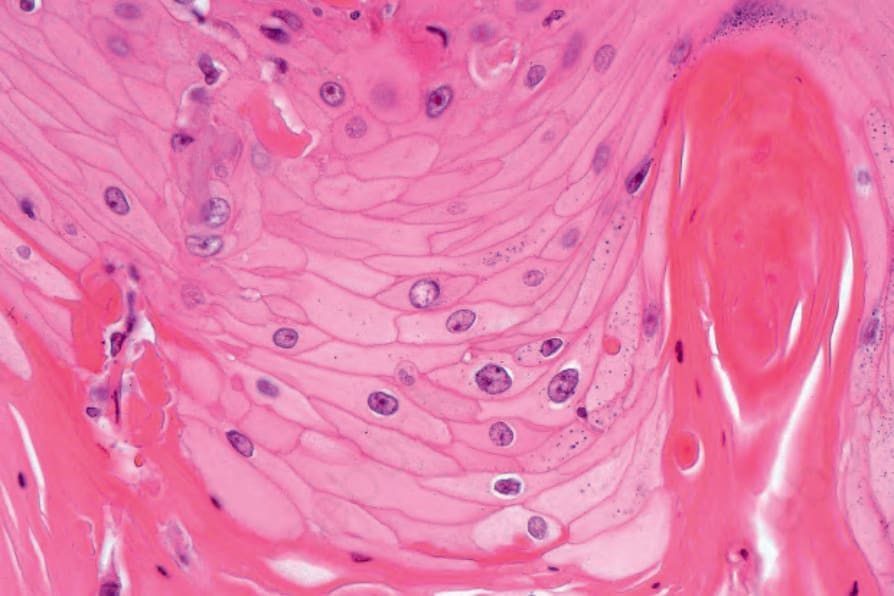
圖 24-184:角化棘皮瘤,增生上皮為高分化,上皮蒼白具特徵性。
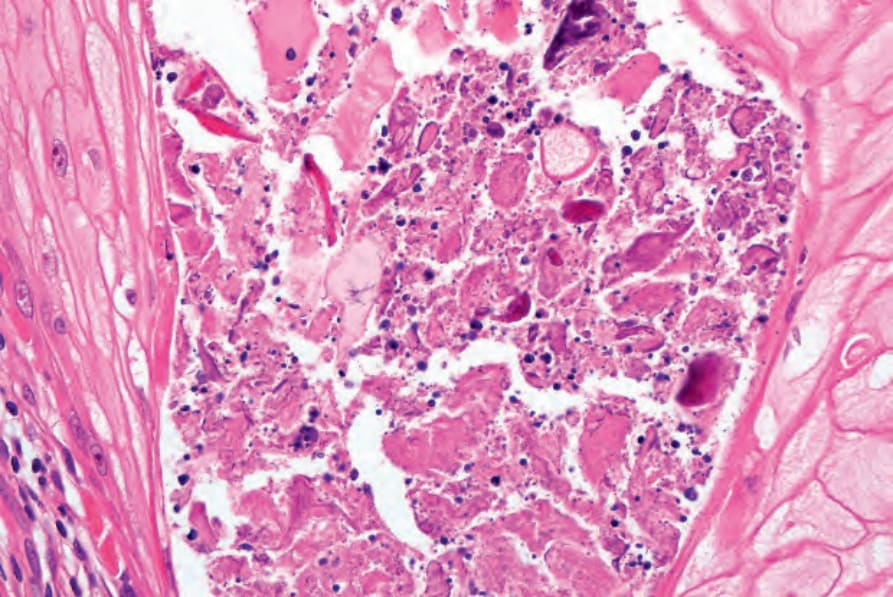
圖 24-185:角化棘皮瘤,此視野的上皮內膿瘍為常見發現。
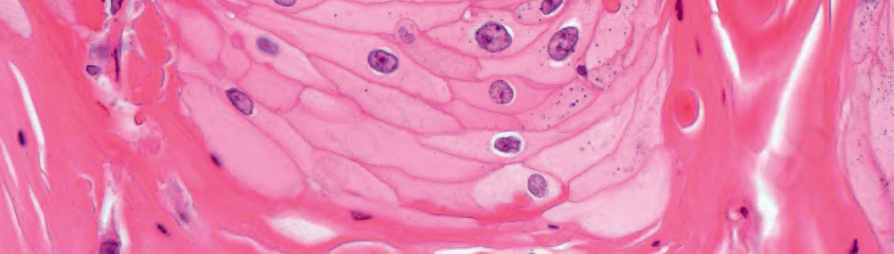
圖 24-186:角化棘皮瘤,有絲分裂一般限於基底上皮層。
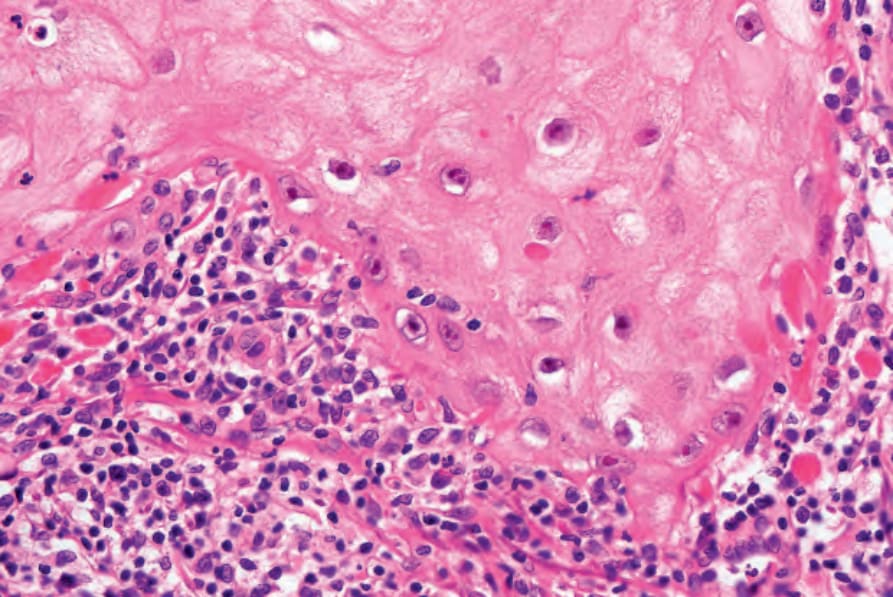
圖 24-187:角化棘皮瘤,常伴混合性慢性發炎細胞浸潤。
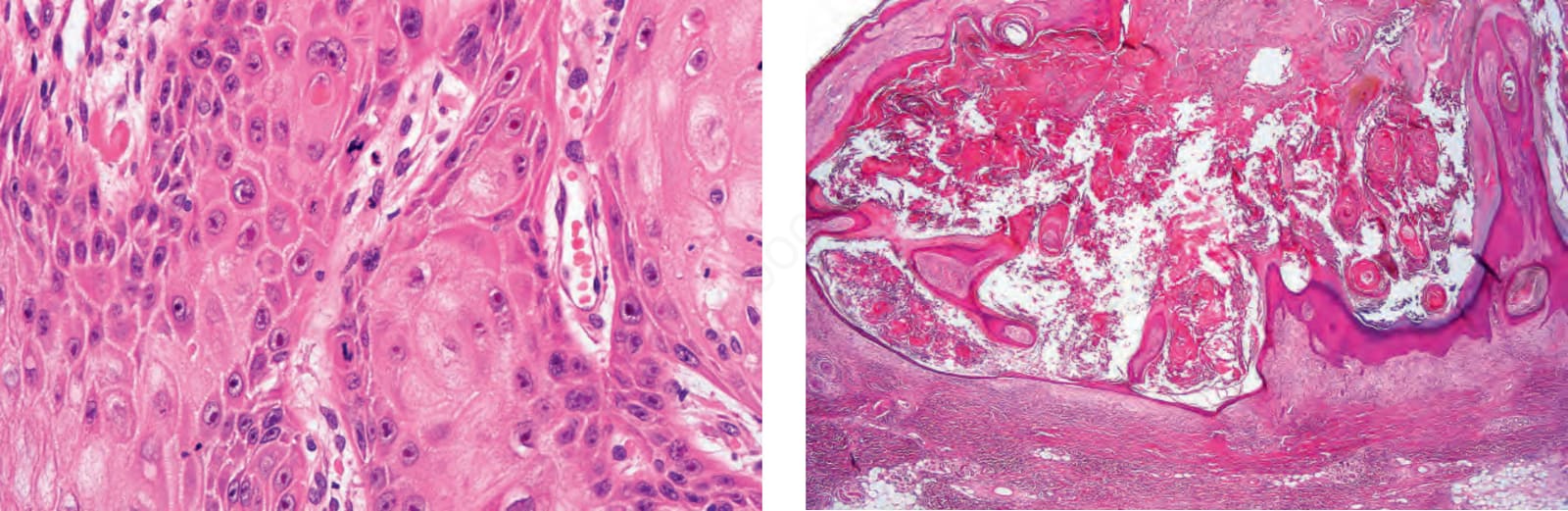
圖 24-188:退化中角化棘皮瘤,角質栓仍在,但上皮增生明顯減少,病灶底部開始變平,注意兩側衣領。
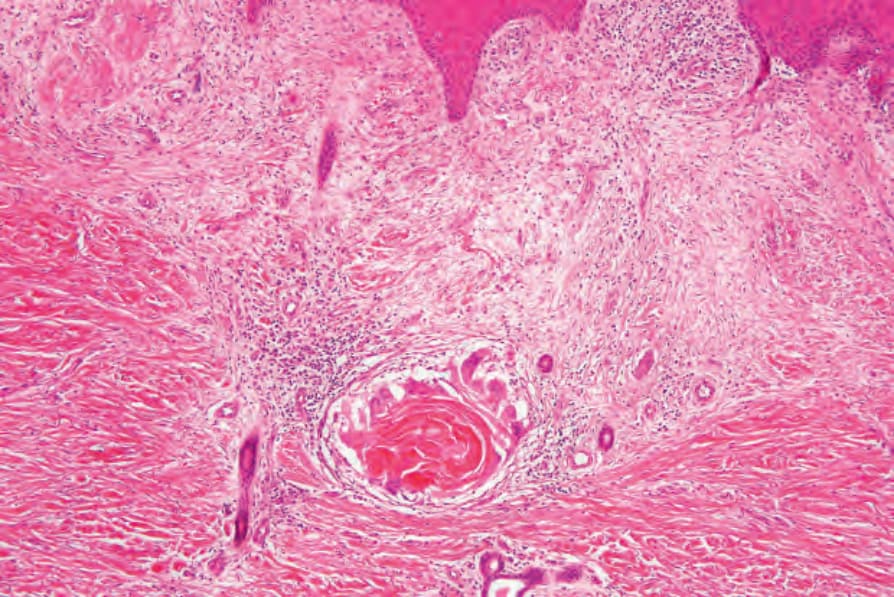
圖 24-189:退化中角化棘皮瘤,病灶基底常見對游離角質的明顯巨細胞反應伴鄰近疤痕。
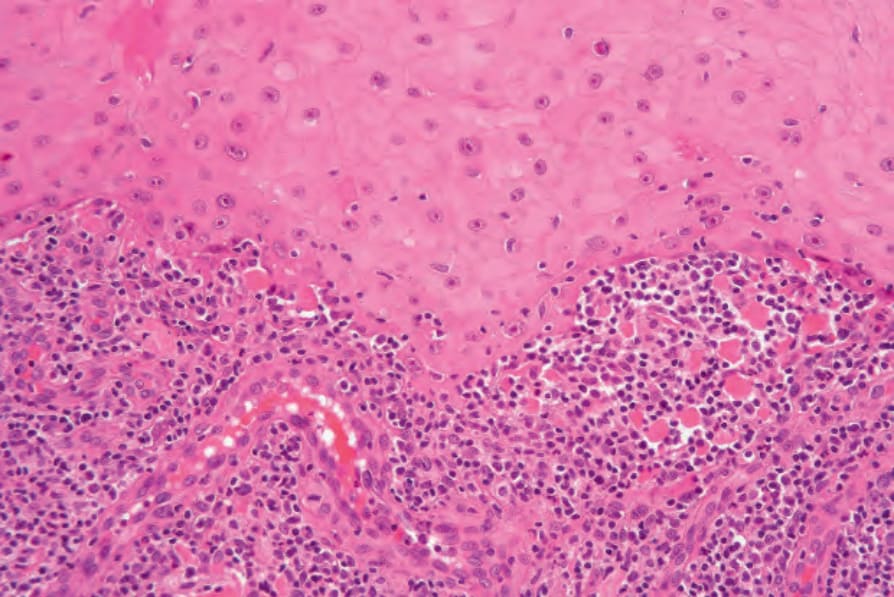
圖 24-190:角化棘皮瘤,此例有明顯界面變化伴顯著類細胞體 (cytoid bodies)。