疾病定義與分類
- 進行性全身性硬化症 (progressive systemic sclerosis) 包含兩大變異:
- 較嚴重的瀰漫型 (diffuse):廣泛皮膚病灶位於 metacarpo- 與 metatarsophalangeal 關節近端,加上內臟(腎、肺、心、食道、腸道)侵犯;常見 anti-Scl-70(anti-DNA topoisomerase)與 RNA polymerase III 抗體,預後一般不佳。
- 局限型 (limited):周邊皮膚硬化、缺乏嚴重全身疾病(食道侵犯、小腸吸收不良、肺高壓除外),預後較佳;與 anticentromere antibody 相關。
- 局限型含 CREST 症候群(calcinosis、Raynaud phenomenon、esophageal dysfunction、sclerodactyly、telangiectasis)。
- 估計發生率約每年每百萬人口 20 新例;女性優勢 (3–4:1),多於第四、五、六十年發病。Juvenile systemic sclerosis 約佔病人 3%。
臨床特徵 (Clinical Features)
- 局限型:手部早期水腫(非凹陷性、雙側對稱)、香腸狀手指,後續皮膚增厚緊繃、手指漸尖、末端指骨吸收;皮膚呈光亮、平滑、蠟樣外觀。進展可有 beaked nose、唇變薄、口周溝紋與張口受限。
- 血管變化:周邊壞疽、手指自截、Raynaud phenomenon(限局型與瀰漫型皆常見)。診斷特徵為甲褶微血管喪失與殘餘微血管擴張。
- CREST:telangiectases 常達數百個,影響手指、手、臉、舌與黏膜。
- 全身性硬化症整體死亡率高,5 年存活率 34% 到 73%;皮膚增厚改善與存活改善相關。
- 色素過度沉著(褐至古銅色)伴局部色素減退,呈「salt and pepper」外觀。
系統侵犯 (重點數值)
- 肺:interstitial pneumonitis(最常見死因)、bronchiolitis(約 13% 到 25% 病人,多無症狀)、pulmonary hypertension(limited 型較常見)。Dyspnea 為近 60% 瀰漫型病人特徵。
- 心:多數病人有亞臨床心臟侵犯;屍檢系列中超過 50% 有心包炎與積液;心肌纖維化見於高達 70% 屍檢病人。
- 腎:‘scleroderma renal crisis’ 約見於 10% 病人,高死亡率;ACE inhibitors 顯著降低死亡率。Anti-RNA polymerase III 抗體見於約三分之一的「scleroderma renal crisis」病人。
- 胃腸:臨床相關病灶見於高達 50% 病人;食道侵犯、逆流、狹窄、Barrett esophagus。
- 鈣質沉著症 (calcinosis cutis) 為 dystrophic 型,因 hydroxyapatite 結晶沉積;phalangeal 再吸收合併 calcinosis cutis 據稱為全身性硬化症的特徵性表現。
致病機轉 (Pathogenesis)
- 病因與精確機轉未明;核心為血管內皮細胞損傷及其後果,與膠原合成異常。
- 多種細胞激素涉入:TGF-β 與 IL-4 增加纖維母細胞增生與膠原合成;IL-17、IL-6、IL-13 等促纖維化與血管異常。
- 纖維化由 type I、III、V、VI 膠原沉積造成,伴過量 fibronectin;type VII 膠原也增加;早期病灶亦含過量 glycosaminoglycans(dermatan sulfate、chondroitin 4- 與 6-sulfate)。
自體抗體 (Autoantibodies)
- Anticentromere antibody:幾乎對全身性硬化症具特異性,尤其 limited 型;通常見於較輕、預後較佳病人。
- Scl-70 (anti-DNA topoisomerase):見於 20% 到 60% 病人,尤其 diffuse 型;高度特異,與嚴重全身侵犯(肺間質纖維化)及不良預後相關。
- 抗核仁型 (nucleolar) 抗體見於 7% 到 46% 病人,非特異;反應對象包括 U3-RNP (fibrillarin)、RNA polymerase I、Th ribonucleoprotein、PM-Scl(見 Table 17.10)。
診斷分類標準
- ARA 主要標準為近端硬皮 (proximal scleroderma)(敏感度 91%、特異度超過 90%);次要標準:sclerodactyly、指尖點狀凹陷瘢痕/末端指墊組織喪失、雙側基底肺纖維化。主要或兩項次要標準有 97% 敏感度、98% 特異度。
- 2013 ACR/EULAR 分類採計分制,總分 ≥ 9 分診斷為確定全身性硬化症;手指增厚延伸至 metacarpophalangeal 關節近端即為充分標準(9 分)。新標準敏感度 98.3%(vs. 1980 年標準 88.3%)。
組織病理特徵 (Histopathology)
- 主要特徵為纖維化(scarring)。早期水腫期與 scleredema of Buschke 難以區分。
- 真皮因寬大、延長、腫脹的膠原束增厚,常與表皮平行;纖維邊界不清呈均質外觀;纖維化常侵犯皮下,故脂肪細胞被併入真皮。汗腺等附屬器萎縮。
- 血管(尤其指動脈)內皮腫脹、內膜增厚、中膜肥厚,後可玻璃樣化;動脈呈明顯內膜增厚(‘onion skinning’)。
- 早期病灶有淋巴球、組織球與少數漿細胞的慢性發炎浸潤,T-helper 細胞為主。
- 通常無法以組織學區分 localized scleroderma (morphea) 與全身性硬化症,但局部型表皮通常正常、血管變化較輕、發炎浸潤常較重且影響網狀真皮。
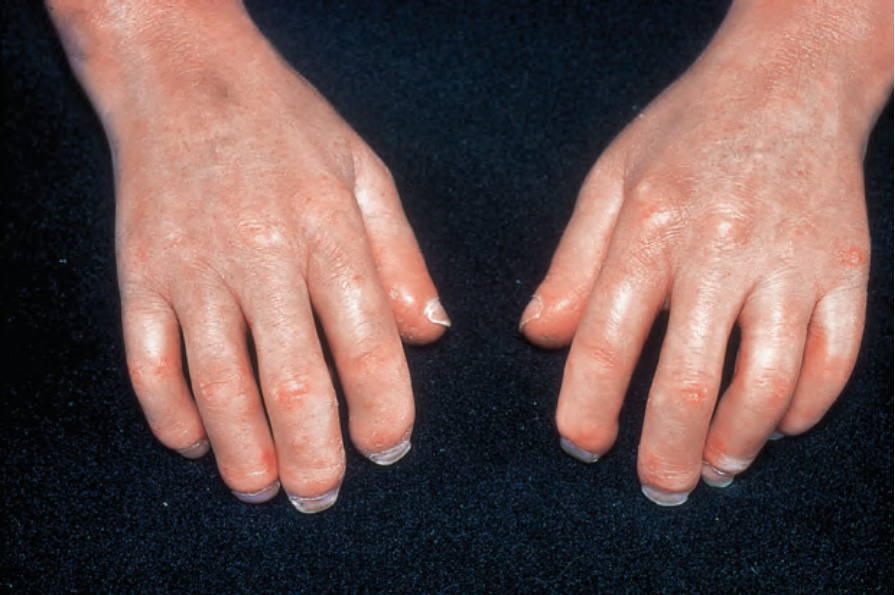
圖 17-82:全身性硬化症:早期顯示特徵性腫脹、香腸狀手指。
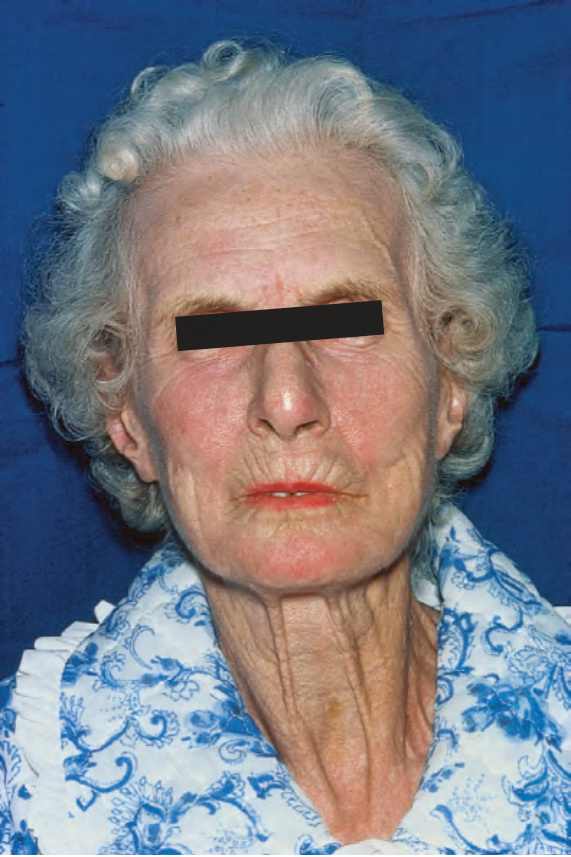
圖 17-88:全身性硬化症:注意變薄的唇與特徵性放射狀溝紋。
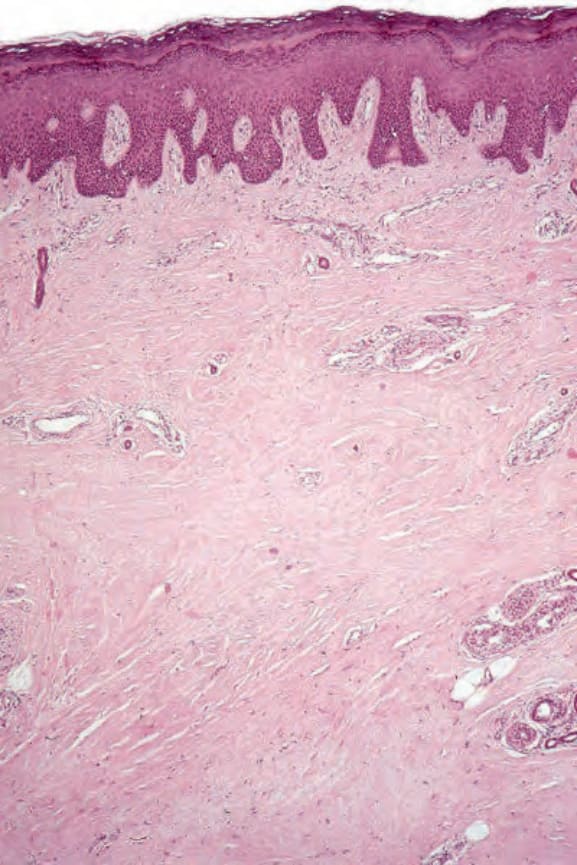
圖 17-96:全身性硬化症:肢端皮膚掃描視野顯示真皮纖維化。
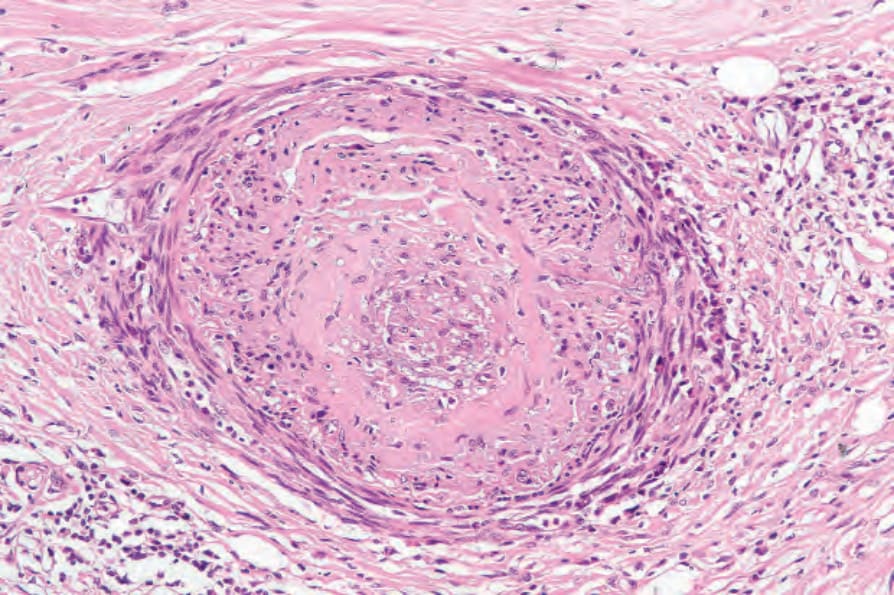
圖 17-98:全身性硬化症:嚴重血管侵犯,內膜纖維化與管腔阻塞,周圍慢性發炎與纖維化。

表 17-7:美國風濕病學會 (ARA) 硬皮病分類指引。
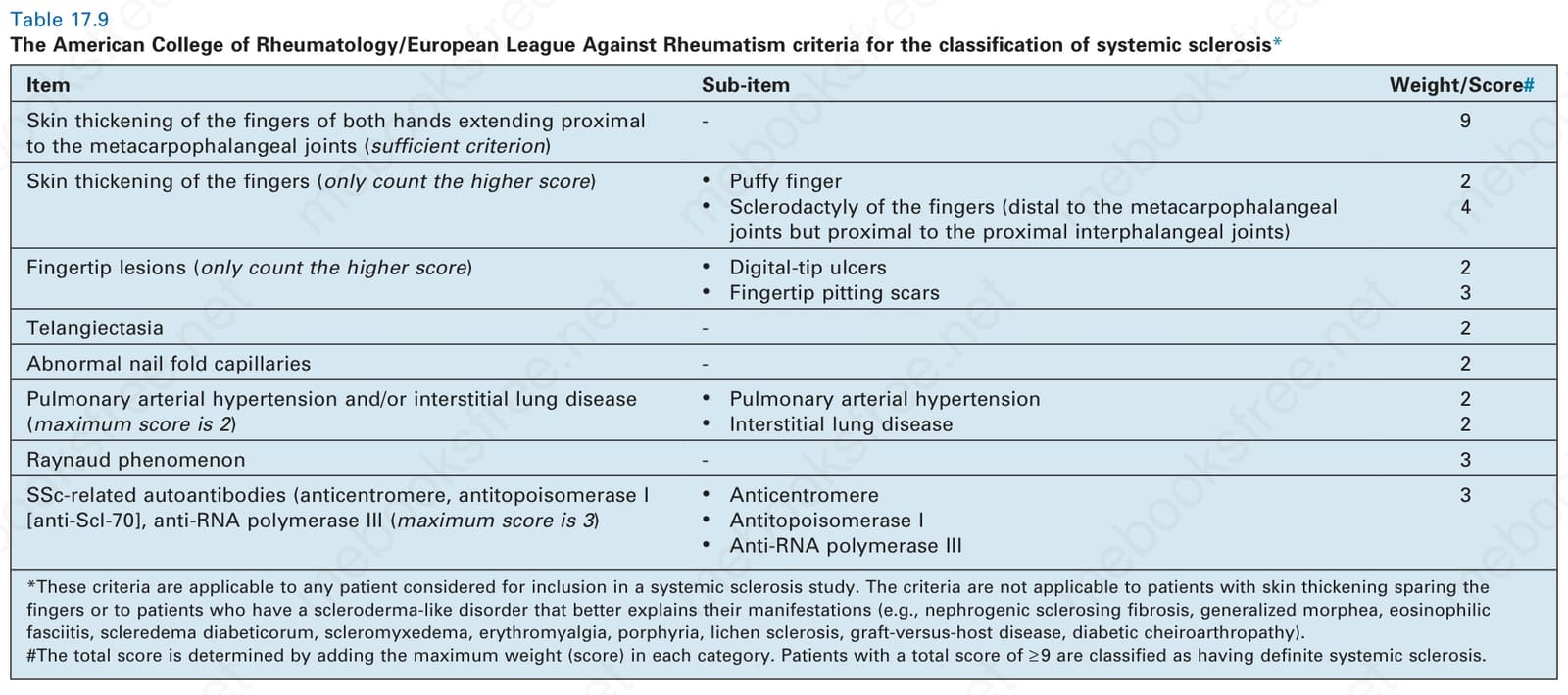
表 17-9:ACR/EULAR 全身性硬化症分類標準。