Systemic sclerosis
Systemic sclerosis
Clinical features Progressive systemic sclerosis includes two major variants:1–5
• In the more serious diffuse form, patients have widespread cutaneous lesions proximal to the metacarpo- and metatarsophalangeal joints (proximal scleroderma) in addition to involvement of the internal viscera, particularly the kidneys, lungs, heart, esophagus, and intestinal tract. The illness often has an acute onset with fatigue, weight loss, arthralgia, and carpal tunnel syndrome. Tendon friction rubs are characteristic. Anti-Scl-70 (anti-DNA topoisomerase) and RNA polymerase III antibodies are often present, and the outlook is generally poor.
• The other major variant is associated with limited peripheral cutaneous sclerosis and an absence of severe systemic disease except for esophageal involvement, small intestinal malabsorption, and pulmonary hypertension; it usually has a better prognosis.2,6 This form is associated with an anticentromere antibody. Systemic sclerosis has also occasionally been recorded in the absence of cutaneous manifestation (sine scleroderma variant), and overlap syndromes have been described.7,8 Limited scleroderma also includes the CREST (calcinosis, Raynaud phenomenon, esophageal dysfunction, sclerodactyly, telangiectasis) syndrome (Thibierge-Weissenbach syndrome, acrosclerosis) where cutaneous disease expression is restricted to the fingers and toes (sclerodactyly) and face (see below).9 Other generalized variants include sclerodermatomyositis, MCTD, and the chemically induced scleroderma-like syndromes.
797 Systemic sclerosis
Because of the variety of systems that can be affected in systemic sclerosis, patients may be primarily under the care of dermatologists, rheumatologists, nephrologists, or other specialists, resulting in difficulties in determining the exact incidence of the disease; it is estimated to be in the order of 20 new cases per million of the population per year.10 In a large series, the diffuse and limited forms were equally common.11 About 10% were classified as overlap syndromes. The disease occurs more frequently in families and it is regarded as the strongest risk factor identified for this condition.12 However, the absolute risk for each family member is low.12 Systemic sclerosis is associated with a marked female predominance (3–4 : 1); although any age group may be affected, patients most often present in their fourth, fifth, and sixth decades.13,14 Young black females constitute a definite subset with a particularly high risk. Occasional familial instances, usually in children, have also been documented.10 Juvenile systemic sclerosis represent about 3% of systemic sclerosis patients.15 No sex predilection is seen before the age of 8 years.15 In contrast to adult-onset systemic sclerosis, juvenile systemic sclerosis is associated with higher incidence of overlap syndromes, especially with polymyositis/dermatomyositis, different sets of antibodies in the serum (anti-PM-Sci and anti-U1-RNP), and improved survival.16–18 Cardiopulmonary diseases have predictive value for survival in juvenile systemic sclerosis.16,17
due to ischemia, show pulp atrophy and absorption of the terminal phalanges, with the fingertips often not protruding beyond the free margin of the nails (Figs 17.83–17.85). The latter may show longitudinal ridging and brittleness or may even be shed. The affected skin has a very characteristic appearance, being shiny, smooth and rather waxy. Patients often have markedly diminished mobility of their hands and feet, and flexion contractures are common (Fig. 17.86). In advanced disease, many patients acquire a dramatically rigid, expressionless face with beaked nose, thinned lips, and perioral furrowing and wrinkling, and an inability to open the mouth fully (Figs 17.87 and 17.88).21 Tightness of the lower eyelids may also be noticed, and the forehead can appear smooth and free of creases.2 Ulceration is a common complication, particularly where taut skin is stretched over bony prominences susceptible to trauma (Fig. 17.89).
Systemic sclerosis has a high overall mortality, 5-year survival rates varying from 34% to 73%.1 It has been demonstrated that improvement of skin thickening is associated with improved survival.19,20 Older patients and males generally fare worst.
Limited cutaneous systemic sclerosis In the limited variant, the cutaneous manifestations, which often initially affect the hands, include early edematous, sclerotic, and late atrophic stages.1 The edema is characteristically nonpitting, bilateral, and symmetrical. The fingers are commonly described as having a sausage-like appearance (Fig. 17.82). The face, forearms, feet, and legs are sometimes affected. As the edema subsides the skin becomes thickened and tight and is bound down to the subcutaneous tissues. Typically, the fingers become tapered and,
Vascular changes are common and include peripheral gangrene, digital autoamputation, and Raynaud phenomenon.21 The last occurs so frequently (in both limited and diffuse forms) that it is often taught that a patient who has it must be presumed to have systemic sclerosis, until proven otherwise. In limited cutaneous systemic sclerosis, Raynaud phenomenon may precede the onset of cutaneous lesions by many years in a large proportion
798 Idiopathic connective tissue disorders
of patients.10 A useful diagnostic feature of systemic sclerosis is loss of many of the nail fold capillaries and dilatation of the remainder. It has recently been demonstrated that the nail fold capillaroscopy abnormalities correlate with diffuse form of systemic sclerosis, severity of cutaneous involvement, number of affected tracts, and the presence of anti-Scl-70 antibodies.22
The cutaneous changes in CREST syndrome are located predominantly distal to the metacarpophalangeal joints, although the dorsum of the hands and mouth can sometimes also be affected. The inflammatory stage is persistent in the CREST syndrome.
Raynaud phenomenon, either alone or with swollen puffy fingers, is by far the most common mode of presentation and telangiectases tend to be much more numerous (often numbering hundreds) than in patients with diffuse systemic sclerosis. They particularly affect the fingers and hands,
799 Systemic sclerosis
face, tongue, and mucous membranes (Figs 17.90–17.92). The telangiectasias seen in this variant of scleroderma may be difficult to distinguish from those seen in hereditary hemorrhagic telangiectasia.23 The esophageal dysfunction is identical to that seen in the diffuse variant, but it tends to be more severe and affects the majority of patients.
and was more common in the limited cutaneous variant of systemic sclerosis.28 More than one of the associated autoimmune conditions may coexist with CREST syndrome.26 It has been suggested that patients with coexistent primary biliary cirrhosis have a distinctive subset of the disease, which tends to be milder and have a better prognosis.29 An exceptional case of antimitochondrial antibody-positive primary biliary cirrhosis developing after an acute myocardial infarction has been reported.30 Although clinically significant primary biliary cirrhosis develops in 2.5% of systemic sclerosis patients, a recent study has demonstrated the presence of antimitochondrial antibodies in 15% of systemic sclerosis patients.31 Overlap syndrome between primary biliary cirrhosis and autoimmune hepatitis has also been found in systemic sclerosis.32 A multicenter French-Italian study has found coexistence of systemic sclerosis and autoimmune disease in 21% of the patients, with Sjögren syndrome (12%) and thyroiditis (6%) being the most frequent associations.33 Systemic sclerosis patients with associated autoimmune conditions appear to follow a milder course of the disease.33 Systemic sclerosis is associated with rheumatoid arthritis in about 5% of the patients, and likely represents a distinctive disease subset associated with HLA-DR3, HLA-DR7, HLA-DR11, and HLA-DRw53.34 In a large cohort of over 2000 patients with systemic sclerosis, association with ANCA-positive vasculitis was established in 1.6% of the patients.35 An overlap between
The value of the designation ‘CREST syndrome’ has, however, diminished considerably since the discovery that many patients with limited cutaneous disease fail to fulfill all of its criteria and the observation that ‘CREST’ manifestations may be seen in many patients with diffuse disease.8 For example, there is a variant consisting of digital necrosis, Raynaud phenomenon, and anticentromere antibodies without sclerodactyly.24 The term should probably be abandoned and all patients with limited distal disease classified as a single subtype. However, considering the frequency with which CREST syndrome appears in the current literature, this is unlikely to happen, at least in the foreseeable future. It was originally thought that the limited variant was associated with a relatively benign outcome, but it is now known that if patients are followed for sufficient time a significant proportion will develop severe pulmonary hypertension with its sequelae. There is also an increased risk of Sjögren syndrome, biliary cirrhosis, discoid lupus erythematosus, and thyroid dysfunction, such as Hashimoto thyroiditis and Graves disease.1,25–27 Sjögren syndrome has been found in 14% of patients with systemic sclerosis
800 Idiopathic connective tissue disorders
systemic sclerosis and cryoglobulineic vasculitis was detected in 1.6% of the patients, usually in the presence of hepatitis C virus infection.36
Hyperpigmentation, from light brown to dark bronze, reminiscent of Addison disease, is a frequent manifestation and is often associated with the presence of small foci of hypopigmentation, giving a characteristic ‘salt and pepper’ appearance. The pigmentary changes particularly affect the backs of the hands and forearms, and the upper part of the chest and back. Sometimes the degree of accompanying hypopigmentation is so marked that it resembles vitiligo.37 Exceptionally, abnormalities in skin pigmentation developing in the absence of associated sclerosis can represent an initial manifestation of incipient systemic sclerosis.38
Rare skin presentation of systemic sclerosis is characterized by multiple and immovable small papules or nodules giving the lesion a so-called cobblestone appearance.39 It is believed that this presentation pattern is the consequence of lymphangiectasia due to the obstruction of lymphatic channels by the fibrosing process.39
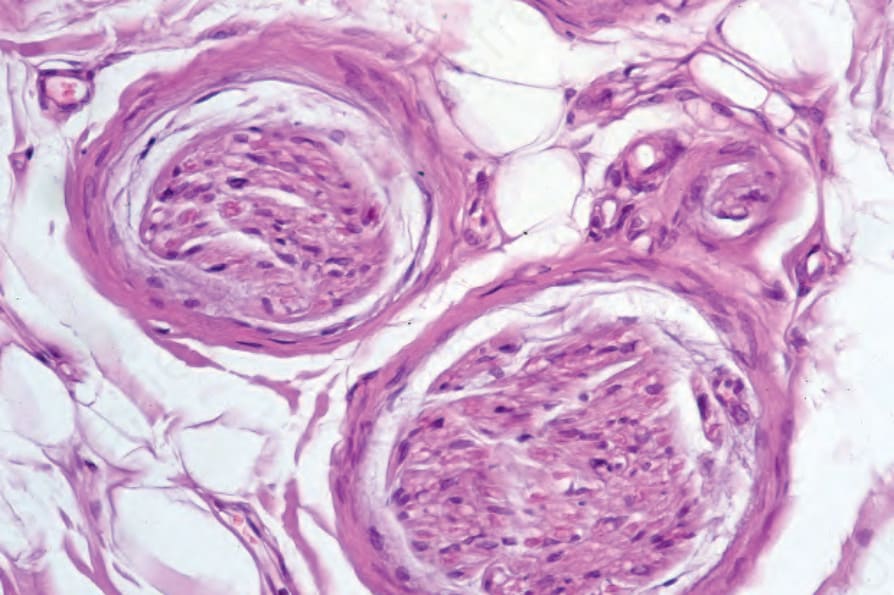
Fig. 17.100 Systemic sclerosis: high-power view.
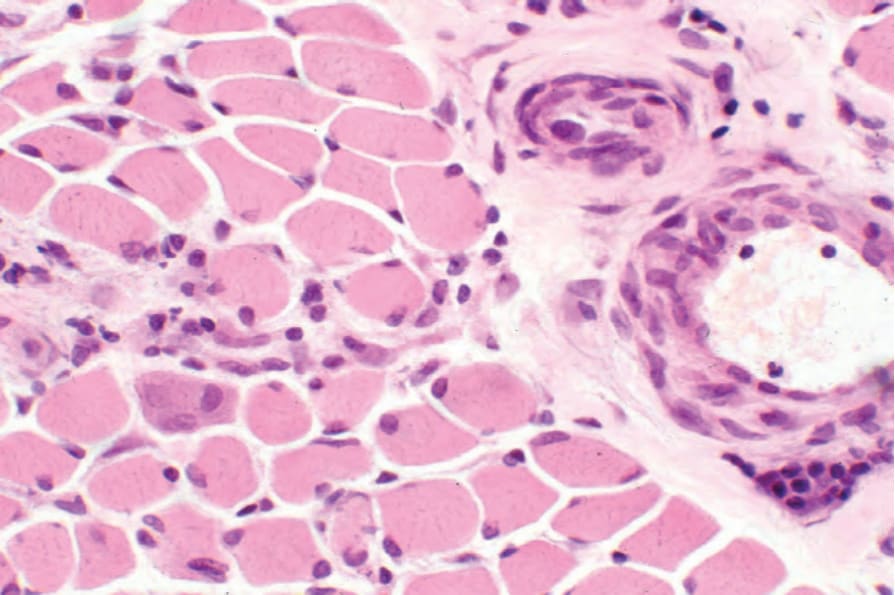
Fig. 17.102 Systemic sclerosis: Note the focal cytoplasmic basophilia on the left side of the field.
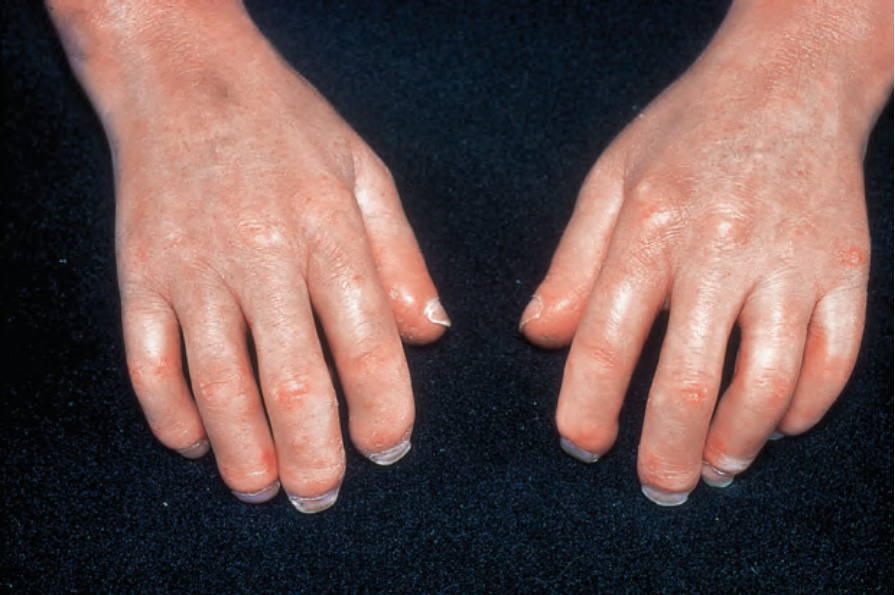
Fig. 17.82 Systemic sclerosis: early stage showing characteristic swollen, sausage-shaped fingers. By courtesy of the Institute of Dermatology, London, UK.
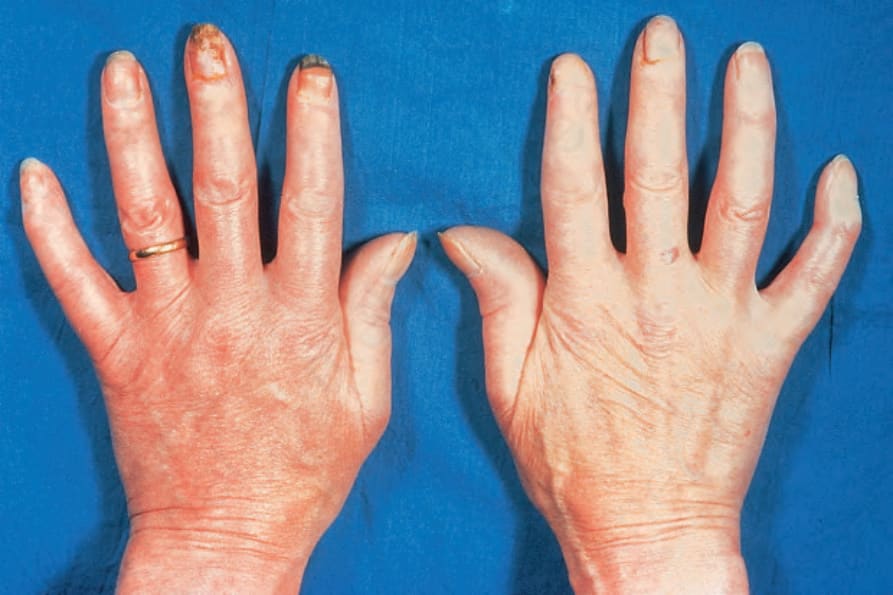
Fig. 17.83 Systemic sclerosis: the fingers are erythematous and shiny and the skin appears slightly bound down. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
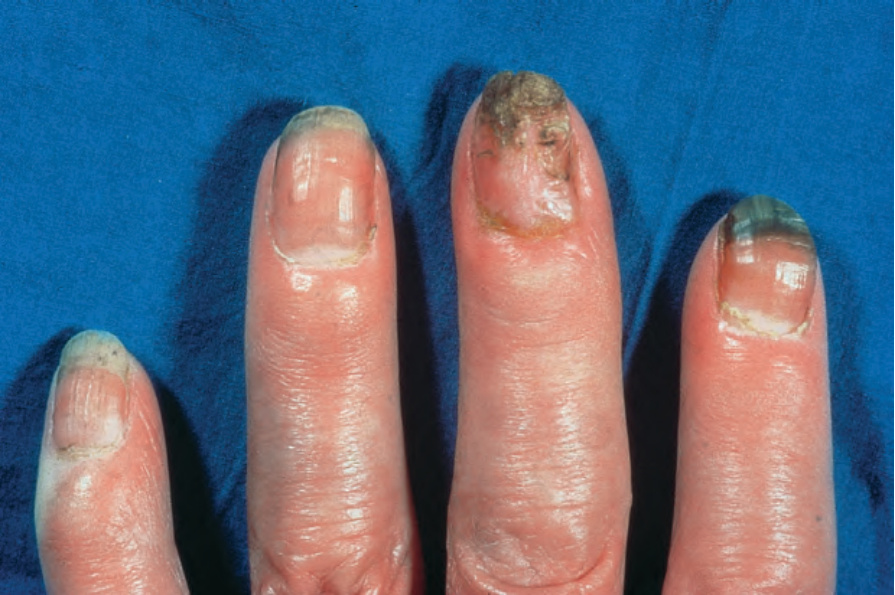
Fig. 17.84 Systemic sclerosis: the fingertips are tapered. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
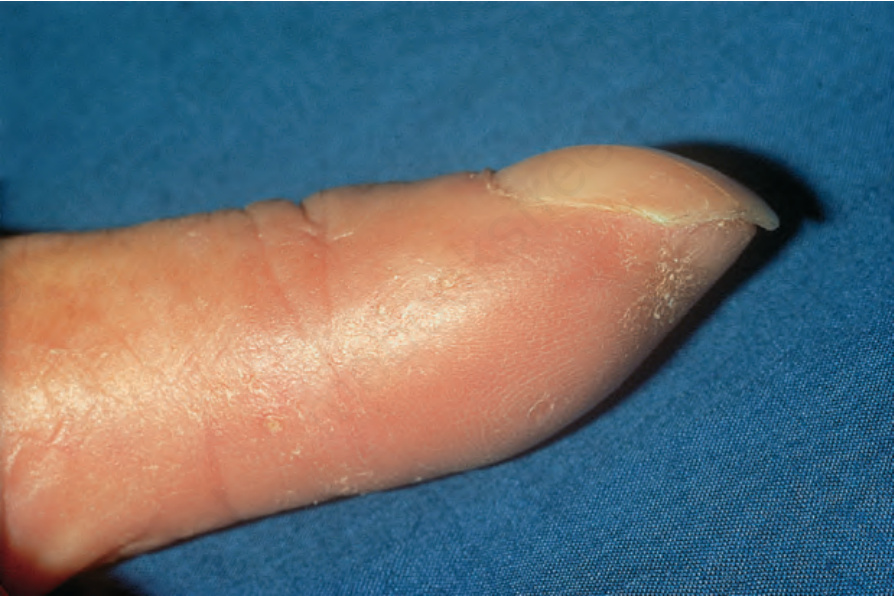
Fig. 17.85 Systemic sclerosis: note the marked atrophy of the fingertip. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
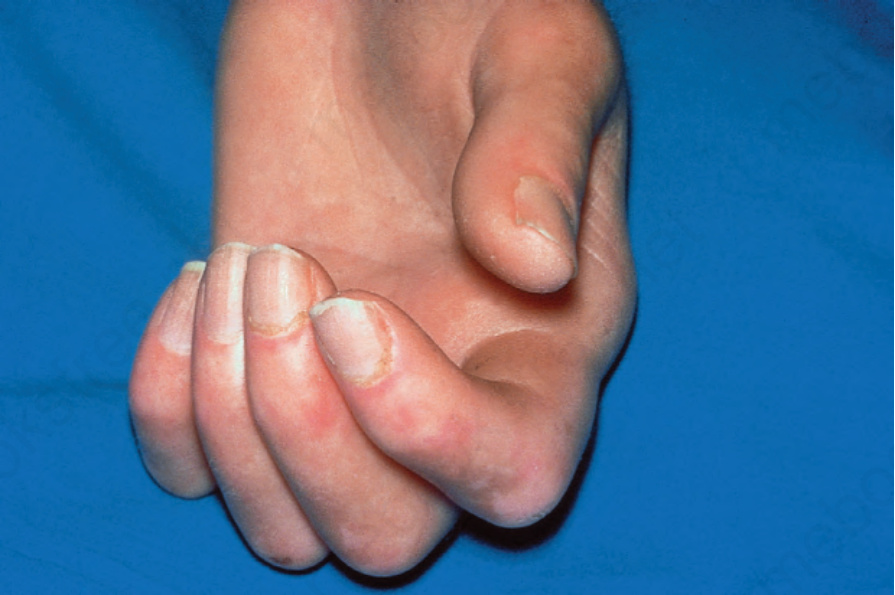
Fig. 17.86 Systemic sclerosis: note the flexion contractures. The skin is bound down and appears atrophic. There is periungual erythema. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
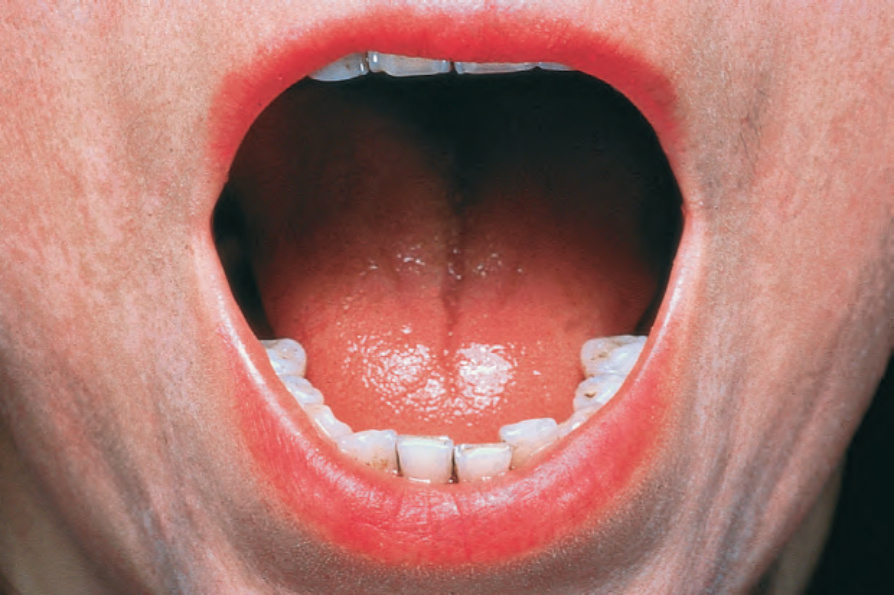
Fig. 17.87 Systemic sclerosis: there is perioral scarring with atrophy. By courtesy of the Institute of Dermatology, London, UK.
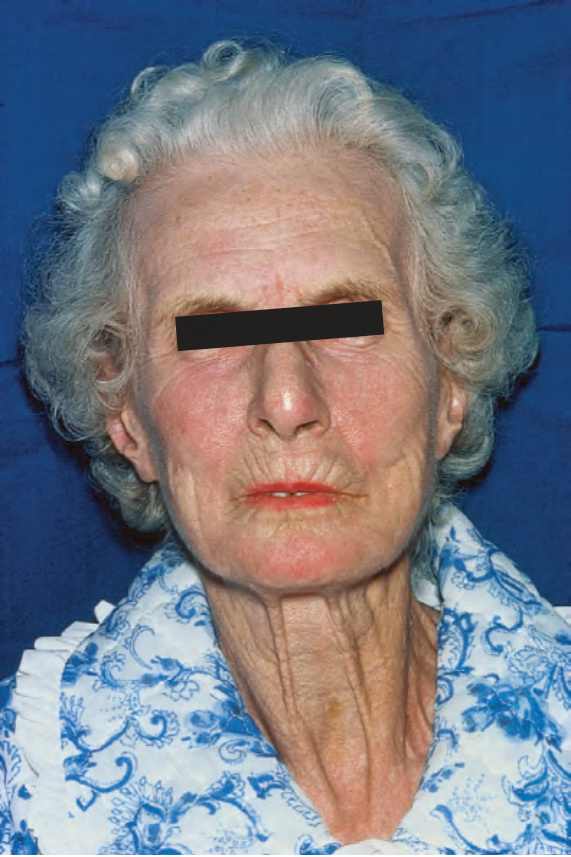
Fig. 17.88 Systemic sclerosis: note the thinned lips and characteristic radiating furrows. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
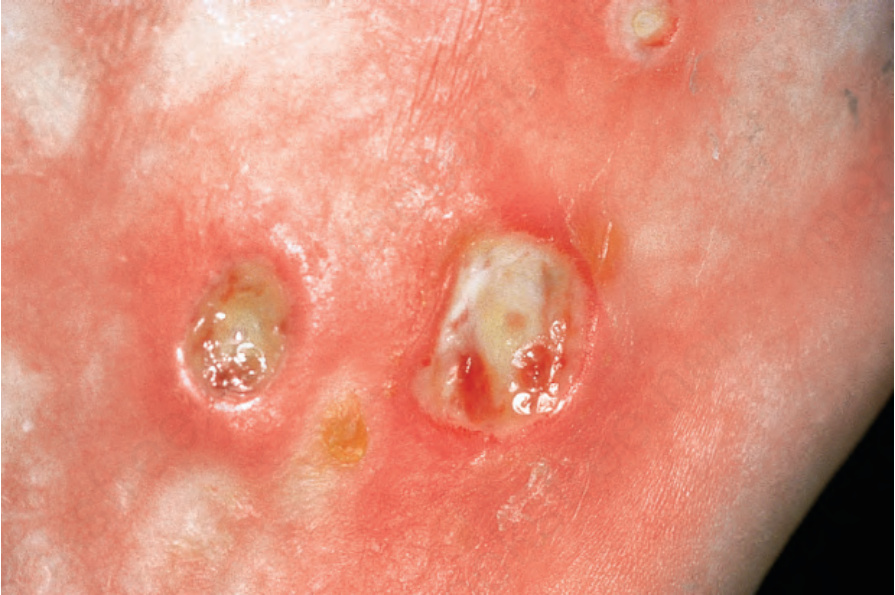
Fig. 17.89 Systemic sclerosis: ulceration is a particularly distressing complication. By courtesy of the Institute of Dermatology, London, UK.
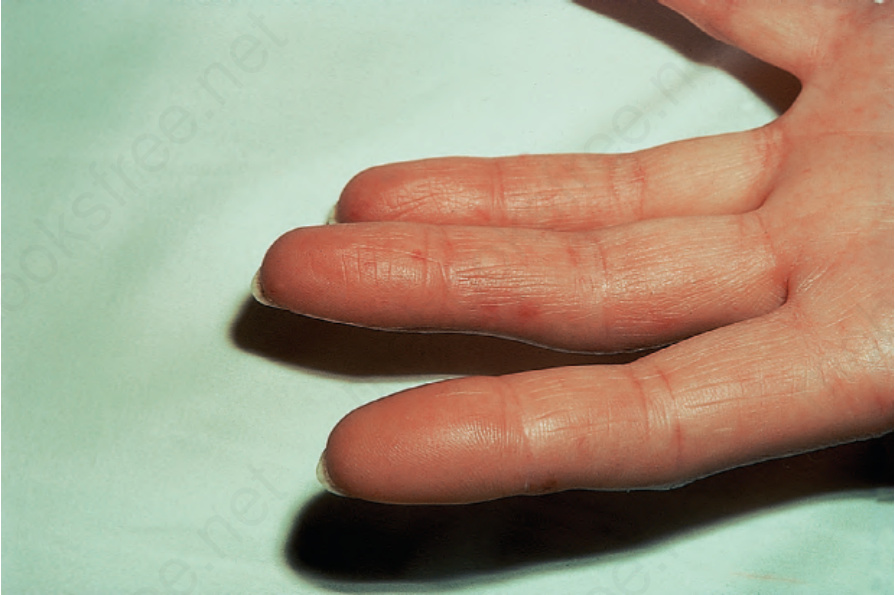
Fig. 17.90 Systemic sclerosis: telangiectasia as seen on these fingers is a common finding. By courtesy of S. Parker, MD, West Middlesex Hospital, London, UK.
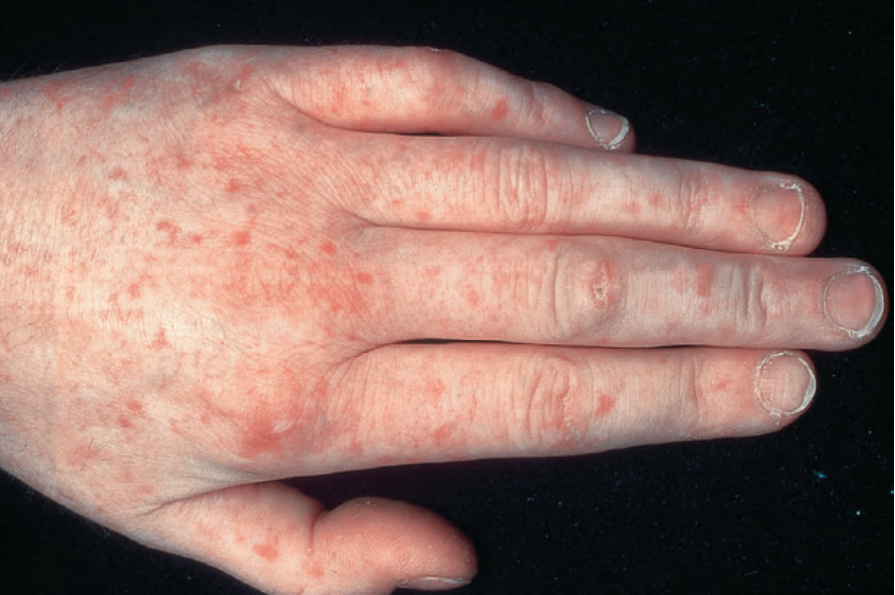
Fig. 17.91 Systemic sclerosis: numerous telangiectases are present. The hand is a commonly affected site. By courtesy of the Institute of Dermatology, London, UK.
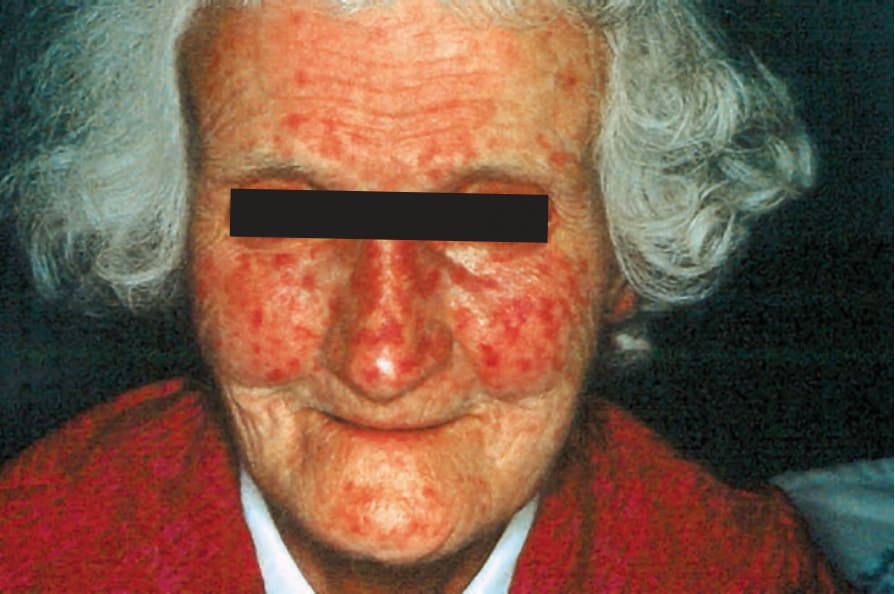
Fig. 17.92 Systemic sclerosis: extensive telangiectasia as seen in this patient is more often a feature of the limited variant. By courtesy of S. Parker, MD, West Middlesex Hospital, London, UK.
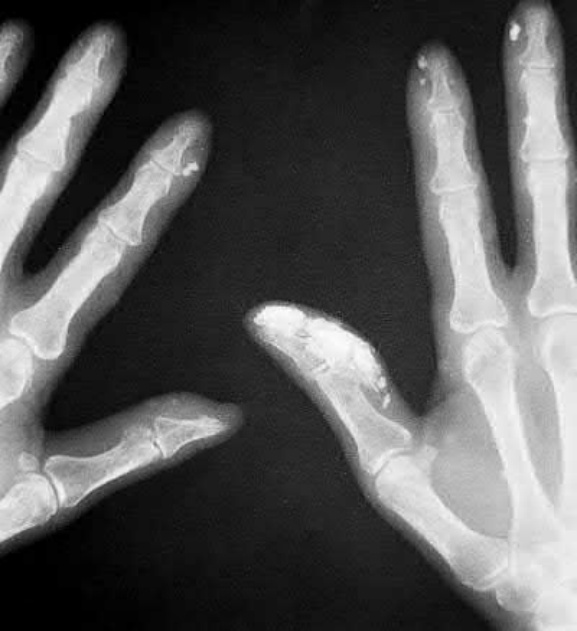
Fig. 17.94 Systemic sclerosis: radiograph demonstrating a more widespread example. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
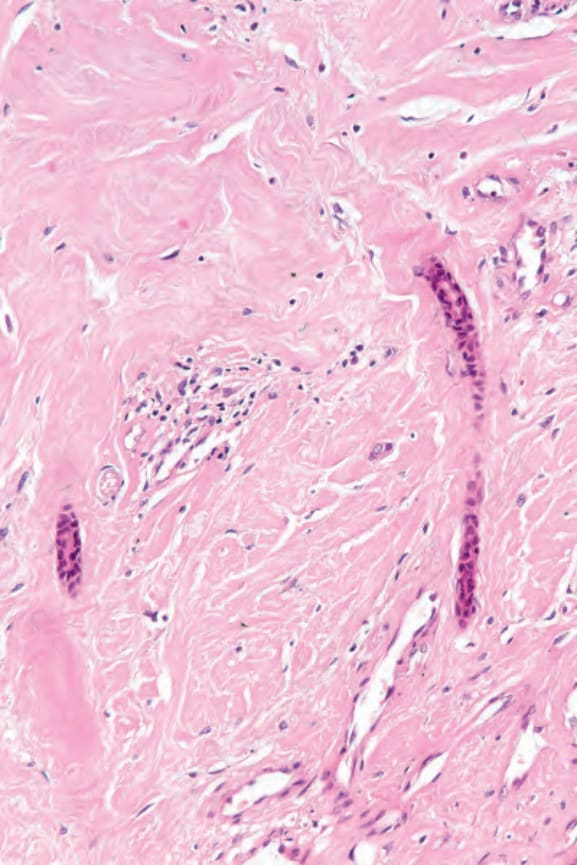
Fig. 17.97 Systemic sclerosis: the dermis is homogenized. Note the compressed eccrine ducts.
early investigation of a patient with systemic sclerosis should establish baseline values of respiratory, cardiac, and renal function so that progress may be accurately monitored.
Clinical involvement of the lung is common and an important cause of morbidity and mortality. It is thought to occur to a greater or lesser extent in most patients.48 There are three major forms: interstitial pneumonitis, bronchiolitis, and pulmonary vascular disease.48
• Pulmonary interstitial involvement results in shortness of breath on exertion and a nonproductive cough. Dyspnea is a feature in almost 60% of unselected patients with diffuse scleroderma.49 Symptoms tend to be particularly evident in patients who have associated Sjögren syndrome.48 Interstitial lung disease is the commonest cause of death in systemic sclerosis. There is evidence suggesting an increased risk of pulmonary fibrosis in patients with the haplotype HLA-DR3/-DRw52A and anti-Scl-70 antibody.50,51
• Bronchiolitis is evident in approximately 13% to 25% of patients, but this is usually asymptomatic.48
• Pulmonary hypertension, which may be a primary manifestation of systemic sclerosis or develop secondary to interstitial fibrosis, is more common in patients with the limited form of the disease.52,53 A study has found that the postmenopausal state with or without the presence of HLA-B35 is the main risk factor for the development of pulmonary hypertension.54
The cutaneous changes are often associated with the development of calcinosis cutis, particularly in females.2,40,41 It is dystrophic in type and is due to hydroxyapatite crystal deposition. Patients have no abnormalities of calcium and phosphorus metabolism and their serum alkaline phosphatase levels are normal. The sites of calcium deposition particularly include the metacarpophalangeal joints of the thumbs and the fingertips, although the extensor aspect of the forearms, the buttocks, the olecranon bursae, and the prepatellar region may also be affected (Figs 17.93 and 17.94). The deposits are often exceedingly painful and commonly associated with ulceration and leakage of white granular calcified debris. The combination of phalangeal reabsorption and calcinosis cutis is said to be pathognomonic of systemic sclerosis.42 Calcinosis cutis has also been reported in patients with systemic sclerosis sine scleroderma.43
Pulmonary radiographs typically show bilateral basal fibrosis, either as diffuse mottling or linear infiltrates; cyst formation (‘honeycomb lung’) is a not uncommon feature.
The cardiac, renal, peripheral nervous, gastrointestinal, and skeletal systems are also involved:
• The majority of patients have subclinical primary cardiac involvement.55 Cardiac involvement may present as dyspnea on exertion, paroxysmal nocturnal dyspnea, pericarditis, pericardial effusion, congestive heart failure, arrhythmias, valvular abnormalities, myocardial hypertrophy or, occasionally, atypical angina.55–57 Vasospasm of the small coronary arteries and arterioles has been observed as an early cardiac manifestation.55 Significant cardiac abnormalities including pericarditis and effusion are common pathological findings, being present in more than 50% of cases at autopsy.58 Usually, however, they are asymptomatic. Occasionally, severe acute pericarditis may develop and rare instances of fatal cardiac tamponade have been documented.56 Large pericardial effusions correlate with acute renal failure and are a bad prognostic indicator.58 Patchy myocardial scarring is common, and usually occurs later in the course of systemic sclerosis.55 When severe, myocardial fibrosis is associated with a poor outlook.59 It occurs
Systemic sclerosis is sometimes associated with an erythema nodosum-like panniculitis syndrome and patients may also develop livedo reticularis and atrophie blanche affecting the lower limbs.44
CREST has been documented in association with familial lichen sclerosus, chronic myelogenous leukemia, idiopathic myelofibrosis, and porphyria cutanea tarda.45–47
Diffuse systemic sclerosis In diffuse (progressive) systemic sclerosis, cutaneous lesions are particularly common on the proximal extremities, thorax, and abdomen. The course tends to be progressive and often there is a more severe superficial vascular involvement.2 Skin thickening affecting the trunk indicates a poor prognosis and correlates with extensive systemic involvement.10 Although the cutaneous features cause considerable distress, the systemic manifestations are more important in terms of severe morbidity and potential mortality. The
independently of coronary artery disease and has been described in up to 70% of patients in postmortem series.56 The major coronary arteries are patent and normal (unless there is coexistent atherosclerosis), but the small vessels and arterioles may undergo endothelial and intimal proliferative changes with scarring, resulting in an increased risk of arrhythmia and the consequent sudden death of the patient. A recent study has found association between myocardial perfusion defects, skin thickness, digital ulcers, and esophageal involvement.60
• Renal involvement presenting as ‘scleroderma renal crisis’ is an extremely important complication with high mortality and occurs in approximately 10% of patients with systemic sclerosis.61 The use of ACE inhibitors has, however, significantly diminished the mortality. It is defined as ‘the new onset of accelerated arterial hypertension and/or rapidly progressive oliguric renal failure’.61 Patients develop headache and blurred vision. Seizures are sometimes a feature. The renal failure is commonly asymptomatic and detectable only from abnormal renal function tests, including proteinuria, microscopic hematuria with casts, raised creatinine levels, and hyperreninemia.61 Microangiopathic hemolytic anemia is sometimes present, particularly in normotensive patients.62 Anti-RNA polymerase III antibodies have been detected in about one-third of the patients with ‘scleroderma renal crisis’.63 A minority of patients with systemic sclerosis develop renal pathology other than ‘scleroderma renal crisis’.64 ANCA-related glomerulonephritis has occasionally been reported.65,66
• Peripheral neuropathy may lead to neuropathic ulceration.67 Intrauterine fetal death has been described in pregnant women with the disease.68 Papular and nodular mucinosis has been documented as a presenting sign of systemic sclerosis.69
• Clinically relevant gastrointestinal lesions occur in up to 50% of patients with systemic sclerosis.70,71 Widening of the periodontal space, determined radiographically, is characteristic. Patients frequently have symptoms relating to esophageal involvement including heartburn, dysphagia, and regurgitation. Gastrointestinal reflux is common and patients can develop esophagitis, hemorrhage, stricture, Barrett esophagus (gastric metaplasia), and aspiration.70 Radiographs may show esophageal dilatation and abnormalities of peristalsis. Epigastric fullness is a frequent symptom, which might be related to restricted distension of the gastric antrum.71 Gastric antral vascular ectasia appears to develop earlier in those systemic sclerosis patients who display a rapidly progressive cutaneous disease.72 Systemic sclerosis often involves the small intestine, symptoms ranging from epigastric pain, nausea and vomiting, through to the effects of pseudo-obstruction; a malabsorptive state due to stasis is an important complication. Celiac disease can develop in patients with systemic sclerosis.73 Colonic lesions may result in diarrhea or constipation. Saccular diverticula along the mesenteric border of the colon are characteristic; they sometimes also affect the small bowel.70
• Osteoarticular involvement, presenting with arthralgia or frank arthritis is seen in the majority of patients.74 Joint lesions are usually mild and affect the wrists, hands, knees, and ankles, although a more serious rheumatoid arthritis-like variant has been documented.75 Osteoarthrosis and psoriatic arthropathy-like manifestations have also been described.74 It is important in patients with significant joint manifestations that overlap syndromes and MCTD are excluded. Contractures and ankyloses resulting in immobility are important complications, and osteoporosis is common due to a combination of immobilization and ischemia. The diagnosis of systemic sclerosis may be readily apparent, but early disease, particularly the diffuse form, may clinically mimic a variety of other diseases, for example, scleredema of Buschke. Late graft-versus-host disease (GVHD) and chronic lesions of porphyria cutanea tarda are typically sclerodermatous.76 The American Rheumatology Association has guidelines for classification, of which the major criterion is proximal scleroderma (Table 17.7).77 Minor criteria are:
• sclerodactyly,
• digital pitting scars on the fingertips or loss of substance of the distal fingerpad,
• bilateral basal pulmonary fibrosis.
801 Systemic sclerosis
- Proximal scleroderma is a single major criterion: sensitivity is 91% and
specificity is more than 90%.
2. Sclerodactyly, digital pitting scars of the fingertips or loss of substance
of the finger pad, and bibasilar pulmonary fibrosis contribute further minor criteria in the absence of proximal scleroderma.
3. The major or two more minor criteria are present in 97% of definite
systemic sclerosis patients, but in only 2% of comparison patients with systemic lupus erythematosus, polymyositis–dermatomyositis or Raynaud disease.
*Preliminary clinical criteria for systemic sclerosis exclude localized scleroderma and pseudo-dermatous disorders. Reproduced with permission from Masi, A.T. et al. and the ARA Subcommittee for scleroderma (1980) Arthritis and Rheumatism, 23, 581–590.
If a patient has either the major or two minor criteria, there is 97% sensitivity for definite systemic sclerosis and 98% specificity. These criteria have gained wide popularity, but have the disadvantage of excluding at least 10% of cases where, despite a concrete diagnosis of systemic sclerosis, neither major nor minor criteria are fulfilled. Unclassifiable and overlap syndromes are also excluded. Other classifications have included two, three, and even four subtypes based upon the extent of cutaneous sclerosis (Table 17.8).78 Limited cutaneous systemic sclerosis may therefore involve the hands, feet, forearms, and face, or skin lesions can be absent, whereas in diffuse disease the trunk skin is also involved. In a comparison of classification by two subtypes (diffuse and limited) or three subtypes (diffuse, intermediate, and limited), the latter correlated best with antibody specificity and survival.79
In 2013, the American College of Rheumatism (ACR) and the European League Against Rheumatism (EULAR) proposed a revision of the classification criteria for systemic sclerosis in order to improve the sensitivity, especially in diagnosing early forms of systemic sclerosis and localized cutaneous systemic sclerosis.80 The new 2013 ACR/EULAR classification criteria for systemic sclerosis apply a point system in such a way that a score of ≥ 9 is needed to classify a patient as having a systemic sclerosis (Table 17.9). In short, according to the new criteria, skin thickening of the fingers extending proximal to the metacarpophalangeal joints is regarded as a sufficient criterion (score 9 points) for the diagnosis of systemic sclerosis. Alternatively, seven additional parameters with varying weights are applied:
- skin thickening of the fingers (puffy fingers 2 points or sclerodactyly 4
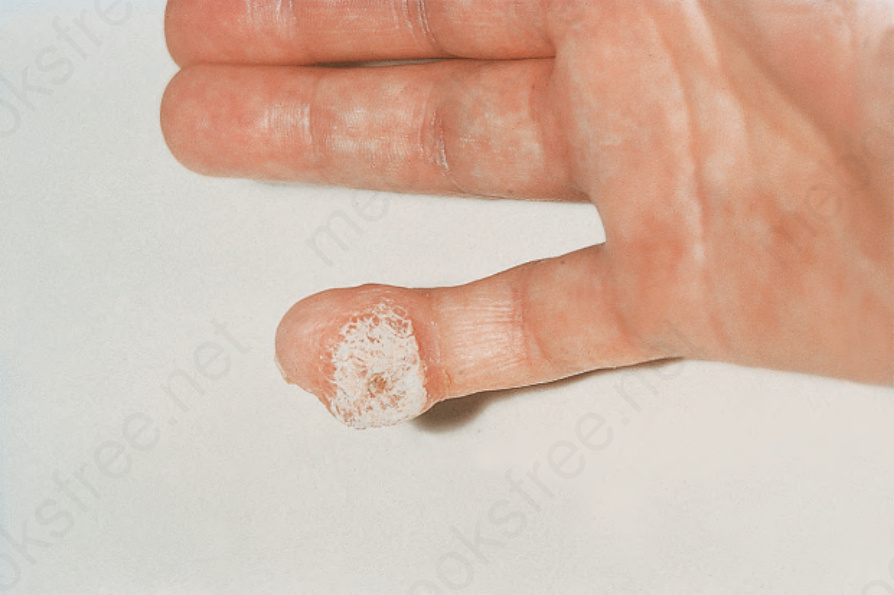
Fig. 17.93 Systemic sclerosis: there is a large calcified nodule on the fingertip. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.

Table 17.7 American Rheumatology Association (ARA) guidelines for the classification of scleroderma*
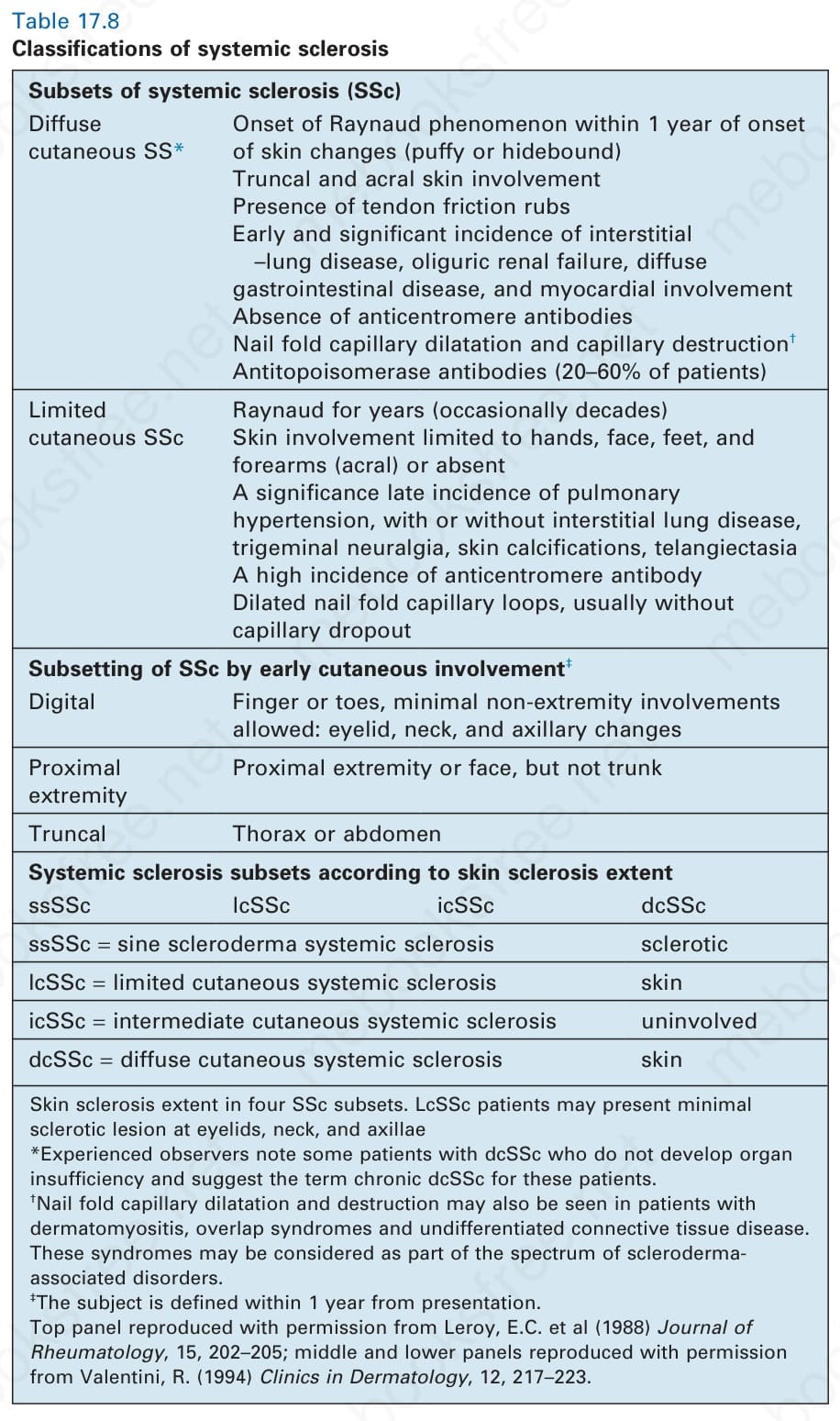
Table 17.8 Classifications of systemic sclerosis
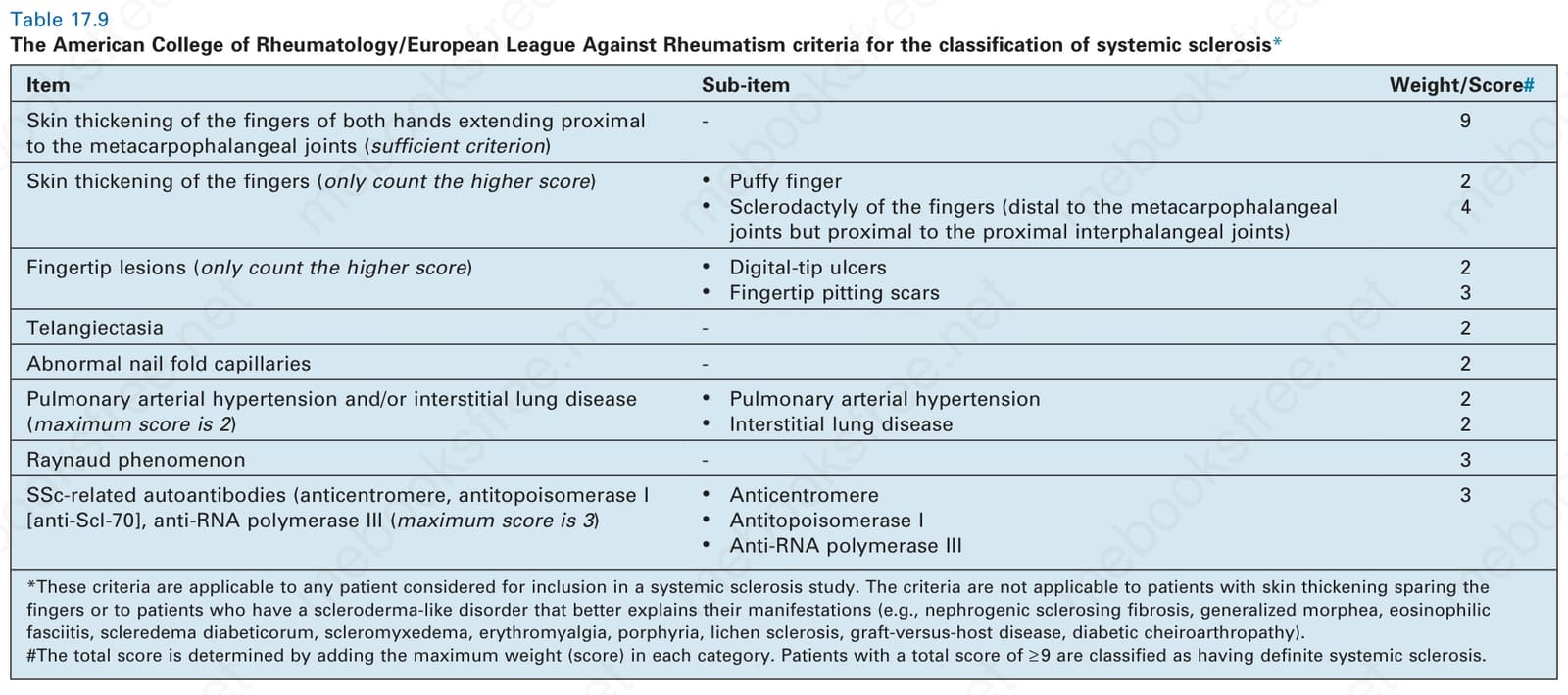
Table 17.9 The American College of Rheumatology/European League Against Rheumatism criteria for the classification of systemic sclerosis*
points, counting the higher score only),
2. fingertip lesions (digital-tip ulcers 2 points or pitting scars 3 points,
counting the higher score only),
3. telangiectasia (2 points),
4. abnormal nail fold capillaries (2 points),
5. interstitial lung disease or pulmonary arterial hypertension (2 points),
6. Raynaud phenomenon (3 points), and
7. systemic sclerosis-specific autoantibodies (3 points).80
By applying the new set of criteria, a large study of 724 systemic sclerosis patients from Canada found the overall sensitivity of the new 2013 criteria to be 98.3% compared to 88.3% for the 1980 criteria, significantly improving the diagnostic accuracy of early systemic sclerosis and localized cutaneous systemic sclerosis.81 In addition, a recent study of 3196 patients with systemic sclerosis revealed the history of digital ulcers to have a predictive value not only for development of new digital ulcer(s), but also for elevated pulmonary arterial pressure, and other possible cardiovascular events, and also represents an independent predictor of decreased survival.82
A number of chemical-induced scleroderma-like syndromes have been described:
• Workers in the vinyl chloride polymerization industry may develop Raynaud phenomenon, acral osteolysis, dermal thickening of the skin of the arms, hands, face, and trunk, and pulmonary and hepatic fibrosis.83–85 Examination of the capillaries of the nail folds reveals abnormalities similar to those seen in systemic sclerosis.
802 Idiopathic connective tissue disorders
Subsets of systemic sclerosis (SSc) Diffuse cutaneous SS*
Onset of Raynaud phenomenon within 1 year of onset of skin changes (puffy or hidebound) Truncal and acral skin involvement Presence of tendon friction rubs Early and significant incidence of interstitial –lung disease, oliguric renal failure, diffuse gastrointestinal disease, and myocardial involvement Absence of anticentromere antibodies Nail fold capillary dilatation and capillary destruction†
incidence of squamous cell carcinoma of the tongue has also been detected.95 Rare associations include basal cell carcinoma, melanoma, nasopharyngeal carcinoma, gastric MALT lymphoma, bladder cancer, cervical cancer, esophageal carcinoma, thyroid cancer, squamous cell carcinoma of the skin, and myelodysplastic syndrome.93,96–100
Pathogenesis and histologic features The etiology and precise pathogenesis are unknown. A complete understanding must take into account vascular changes and abnormalities of collagen deposition and distribution, in addition to the significance of the inflammatory cells that characterize the early stages and their role in the control of fibroblast growth and function.101 Systemic sclerosis has stimulated an enormous research effort, which has resulted in an increased awareness of the multiplicity of factors that may be involved, either singly or in concert, and has also greatly increased our knowledge of the basic processes involved in the mechanisms of collagen synthesis and scarring. The two main areas of investigation have revolved around:
• primary blood vessel endothelial cell damage and its sequelae,
• abnormalities of collagen and its synthesis.102,103
Antitopoisomerase antibodies (20–60% of patients)
Limited cutaneous SSc
Raynaud for years (occasionally decades) Skin involvement limited to hands, face, feet, and forearms (acral) or absent A significance late incidence of pulmonary hypertension, with or without interstitial lung disease, trigeminal neuralgia, skin calcifications, telangiectasia A high incidence of anticentromere antibody Dilated nail fold capillary loops, usually without capillary dropout
Subsetting of SSc by early cutaneous involvement‡
Digital Finger or toes, minimal non-extremity involvements allowed: eyelid, neck, and axillary changes
Inherent to both are the possible initiating and moderating roles of cell-mediated and humoral immunity.
It has long been recognized that many of the features of systemic sclerosis may have an ischemic basis.104 Alterations have been described in capillaries, venules, and arteries, and it has been suggested that the initial injury involves capillary endothelial cells.105 The cause of this is unknown, although a circulating specific cytotoxic substance reactive for endothelial cells has been identified.105,106 It has been suggested that this may represent a protease.107 Interestingly, specimens of early lesions and uninvolved skin have shown ultrastructural evidence of endothelial cell damage combined with decreased uptake of tritiated adenosine and diminished stores of immunodetectable von Willebrand factor, suggesting that the vascular changes may well initiate the connective tissue damage seen in this disease.108
Proximal extremity
Proximal extremity or face, but not trunk
Truncal Thorax or abdomen
Systemic sclerosis subsets according to skin sclerosis extent ssSSc lcSSc icSSc dcSSc
ssSSc = sine scleroderma systemic sclerosis sclerotic
lcSSc = limited cutaneous systemic sclerosis skin
icSSc = intermediate cutaneous systemic sclerosis uninvolved
dcSSc = diffuse cutaneous systemic sclerosis skin
Skin sclerosis extent in four SSc subsets. LcSSc patients may present minimal sclerotic lesion at eyelids, neck, and axillae *Experienced observers note some patients with dcSSc who do not develop organ insufficiency and suggest the term chronic dcSSc for these patients. †Nail fold capillary dilatation and destruction may also be seen in patients with dermatomyositis, overlap syndromes and undifferentiated connective tissue disease. These syndromes may be considered as part of the spectrum of sclerodermaassociated disorders. ‡The subject is defined within 1 year from presentation. Top panel reproduced with permission from Leroy, E.C. et al (1988) Journal of Rheumatology, 15, 202–205; middle and lower panels reproduced with permission from Valentini, R. (1994) Clinics in Dermatology, 12, 217–223.
Although immunoreactants (IgG and complement) have been detected in the walls of renal glomerular capillaries by immunofluorescent techniques, they have not been found in the cutaneous vasculature.108,109 If Raynaud phenomenon is induced in patients with systemic sclerosis, there is a concomitant reduction in both renal and pulmonary blood flow, implying a circulating factor, as yet unidentified.
• Bleomycin therapy may also be associated with sclerodermiform infiltrated plaques and nodules that particularly affect the hands.86 Patients may develop hyperpigmentation, peripheral gangrene, and pulmonary fibrosis.
• A high incidence of systemic sclerosis is found in those who work in coalmines or who have excessive exposure to silica for other reasons.87
• A generalized morphea-like variant with Raynaud phenomenon, esophageal dysfunction, and pulmonary fibrosis has been described following chronic exposure to industrial solvents.88
• A variety of autoimmune diseases have been documented following the use of silicone breast implants. Systemic sclerosis appears to be the most common.89
• Toxic oil and eosinophilia-myalgia syndromes are discussed in the section on eosinophilic fasciitis. Systemic sclerosis is associated with increased risk of malignancies, which develop in between 3.6% and 10.7% of patients.90 Population-based studies have found most frequent association with breast cancer, lung cancer, and hematological malignancies, such as non-Hodgkin lymphoma.91–94 Increased
The dermal capillaries show a variety of ultrastructural changes. The earliest finding is separation of the endothelial cells; this may result in fluid leakage and therefore be responsible, at least in part, for the edema that characterizes the early stages.1 Evidence of more severe damage is manifest by the presence of endothelial cell vacuolation, increased numbers of intermediate filaments, reduction in pinocytotic vesicles and Weibel-Palade bodies, and abnormal endothelial surface cytoplasmic blebs.108
Evidence of endothelial cell injury can be monitored clinically by estimating plasma von Willebrand factor levels.103 Elevated levels of supranormal von Willebrand factor multimers are typically seen in systemic sclerosis and may have pathogenetic significance as they are known to bind to subendothelial tissues, causing platelet aggregation and adhesion with resultant vascular proliferation and thrombosis.103,110 ACE levels have been shown to be reduced in systemic sclerosis and this may also be of value in assessing the presence of endothelial cell damage.102 Increased levels of the endothelial cell-derived peptide, endothelin, which causes vasoconstriction, have been identified.89 Endothelin also has fibroblast mitogenic activity and stimulates the synthesis of collagen.111
The end stage appears as complete destruction of the capillary wall; the nuclei are granular and homogeneous, cell membranes are disrupted, and cytoplasmic contents are found in the capillary lumen and extravascular spaces. Endothelial cell uptake of tritiated adenosine has been shown to be reduced.108 Basement membrane thickening and reduplication is often present and perivascular fibrosis is a common late accompaniment. The end result of vascular damage can be demonstrated most easily by nail fold capillaroscopy. It is likely that the dilatation of the residual vessels represents a compensatory measure. Increased proliferation of the endothelial cells in these residual vessels has been confirmed by tritiated thymidine uptake studies.101
803 Systemic sclerosis
Item Sub-item Weight/Score#
Skin thickening of the fingers of both hands extending proximal to the metacarpophalangeal joints (sufficient criterion)
- 9
Skin thickening of the fingers (only count the higher score)
• Puffy finger
• Sclerodactyly of the fingers (distal to the metacarpophalangeal joints but proximal to the proximal interphalangeal joints)
2 4
Fingertip lesions (only count the higher score)
• Digital-tip ulcers
• Fingertip pitting scars 2 3
Telangiectasia - 2
Abnormal nail fold capillaries - 2
• Pulmonary arterial hypertension
• Interstitial lung disease 2 2
Pulmonary arterial hypertension and/or interstitial lung disease (maximum score is 2)
Raynaud phenomenon - 3
• Anticentromere
• Antitopoisomerase I
• Anti-RNA polymerase III
SSc-related autoantibodies (anticentromere, antitopoisomerase I [anti-Scl-70], anti-RNA polymerase III (maximum score is 3)
3
*These criteria are applicable to any patient considered for inclusion in a systemic sclerosis study. The criteria are not applicable to patients with skin thickening sparing the fingers or to patients who have a scleroderma-like disorder that better explains their manifestations (e.g., nephrogenic sclerosing fibrosis, generalized morphea, eosinophilic fasciitis, scleredema diabeticorum, scleromyxedema, erythromyalgia, porphyria, lichen sclerosis, graft-versus-host disease, diabetic cheiroarthropathy). the total score is determined by adding the maximum weight (score) in each category. Patients with a total score of ≥ 9 are classified as having definite systemic sclerosis.
Arterioles are also involved in the vasodestructive phenomenon, characterized by vessel wall thickening due to a combination of smooth muscle hyperplasia, fibrosis, and the deposition of excessive glycosaminoglycans. Arteries show very marked intimal thickening, which is particularly well seen in the renal arcuate vessels and is often referred to as ‘onion skinning’ due to the concentric lamination. It develops as a consequence of myxoid change, cellular proliferation, and fibrosis.
Most of the inflammatory cells in the skin of patients with systemic sclerosis are CD4+ T cells.
A number of cytokines have been linked to the pathogenesis of systemic sclerosis. Transforming growth factor beta (TGF-β) and IL-4 increase fibroblast proliferation and collagen synthesis and may be important in the induction of fibrosis in this disease. IL-17 is a cytokine secreted by T cells that activates and induces proliferation of fibroblasts and activation of endothelial cells. This cytokine has been demonstrated to be increased in the skin and blood of affected patients, particularly in the early stages of the disease. It activates fibroblasts to secrete the proinflammatory cytokines IL-6 and -8 and to increase surface expression of intercellular adhesion molecule-1 (ICAM-1).112–114 IL-17 also activates endothelial cells to secrete IL-6 and -1 and to express ICAM-1 and vascular cell adhesion molecule-1 (VCAM-1). IL-6 is also capable of inducing proliferation of fibroblasts and collagen synthesis. The combined effects of IL-17 and other cytokines induced by it lead to damage to the microcirculation and to fibrosis in the skin and internal organs. It has been suggested that connective tissue growth factor, the production of which is induced by TGF-β, may play an important role in the pathogenesis of fibrosis.115 It has recently been demonstrated that CD8+ effector T lymphocytes are the source of increased IL-13 in the sera of patients with systemic sclerosis.116 IL-13 has well-known profibrotic activities by direct fibroblast stimulation and, indirectly, by stimulation of TGF-β.117 TGF-β in addition to fibrogenesis also contributes to the development of vascular abnormalities in systemic sclerosis by inducing synthesis of endothelin, which acts as a potent vasoconstrictor and has been implicated in the pathogenesis of ulcers.118
specific, it varies among ethnic groups. Native Americans and Japanese patients have a high frequency of anti-fibrillin-1 antibodies.120
Male cells have been found in multiple organs in women with systemic sclerosis but not in healthy women.121 The migration of fetal cells into maternal circulation and their survival in different organs is known as microchimerism. It is still not clear what role, if any, microchimerism plays in the pathogenesis of systemic sclerosis.
The predominant histologic feature of systemic sclerosis is scarring. Intensive investigations have confirmed the presence of increased quantities of collagen, but as yet the precise pathogenetic mechanism(s) remain uncertain. Increased proline hydroxylase activity and increased uptake of labeled proline, both indicators of active collagen synthesis, have been demonstrated in patients with systemic sclerosis.122 There is typically an elevated level of reducible aldimine cross-links, a feature of newly synthesized collagen.123 Raised serum concentration of the N-terminal propeptide of type III collagen and increased urinary excretion of hydroxyproline have also been documented.103
Cultures of fibroblasts from patients with systemic sclerosis synthesize more collagen than do those from normal controls.124 Although diminished levels of tissue collagenase have been reported, other workers have not confirmed this finding and its significance is therefore uncertain.125 The amino acid composition of the collagen fibers is normal. Electron microscopy of evolving lesions has revealed the presence of immature collagen fibrils, characterized by a narrow caliber (30 nm), immature banding pattern, and double-stranded beaded filaments.1 In the more mature lesion the collagen fibers approach normal thickness (100 nm), but their distribution is highly disorganized. Luse bodies are sometimes a feature.
The fibroblasts in systemic sclerosis are capable of assembling microfibrils but these are unstable (probably due to an inherent defect of fibrillin 1, the extracellular matrix protein) and this may also play a role in the pathogenesis of the disease.119 Interestingly, duplication in the fibrillin-1 gene has been implicated as the cause of tight skin,1 which is an animal model of systemic sclerosis.108 Antibodies against fibrillin are raised in the sera of patients with systemic sclerosis. Although this appears to be highly disease
Patients with diffuse and limited cutaneous systemic sclerosis have increased mean serum levels of soluble CD163 in comparison with the healthy controls.126 It has recently been demonstrated that systemic sclerosis patients with elevated serum levels of soluble CD163 have higher pulmonary arterial systolic pressure than those with normal serum CD163 levels, suggesting the possible role of macrophages in the pathogenesis of systemic sclerosis.126 In contrast, by binding to a TNF-like weak inducer of apoptosis, CD163 may protect against development of digital ulcers, yet contribute to more prominent fibrosis of the skin.127
The serum levels of soluble T-cell immunoglobulin and mucin domain 3 (TIM-3) are higher in patients with diffuse cutaneous systemic sclerosis than in those with limited cutaneous systemic sclerosis and healthy individuals, and positively correlate with the severity of skin sclerosis, especially in the
804 Idiopathic connective tissue disorders
early phase of diffuse cutaneous systemic sclerosis.128 Increased serum levels of TIM-3 have also been associated with cardiac involvement and renal crisis.128
Histologic examination of active lesions often reveals increased numbers of fibroblasts. It has been shown that fibroblasts from the lower dermis synthesize more collagen than do those derived from the upper dermis, suggesting two different populations in systemic sclerosis.129 The fibrosis, which is due to the deposition of types I, III, V, and VI collagen, is accompanied by excessive fibronectin.103,130
on anti-fibrillarin (anti-U3-RNP) autoantibodies confirmed the association with younger age at the disease onset, male gender, Afro-Caribbean descent, higher Rodnan skin score severity index, and myositis, but not with the presence of diffuse cutaneous systemic sclerosis, lung involvement, or differences in survival.137 Anti-U11/U12-RNP antibodies have been demonstrated in about 3% of systemic sclerosis, and have increased risk of pulmonary fibrosis and gastrointestinal involvement.138 Anti-Ku antibodies are detectable in 2.2% of systemic sclerosis and have been related to musculoskeletal abnormalities, such as myositis, arthritis, and joint contractures, as well as fingertip ulcers and telangiectasias.139
Recently, abundant type VII collagen has also been demonstrated within the dermis of involved skin accompanied by elevated expression of TGF- β.130 The latter is known to upregulate the activity of the type VII collagen gene. This finding is of potential importance as type VII collagen distribution is normally restricted to the anchoring fibrils at the dermal–epidermal junction. Increased expression of types I and III collagen mRNA has been demonstrated in cultured fibroblasts from patients with scleroderma.131,132 Systemic sclerosis is characterized by a normal concentration of collagen per unit weight. In contrast, however, there is a greatly increased collagen content per unit surface area.133
Collagen synthesis has a negative feedback control. Therefore, following cleavage of the amino terminal of the procollagen molecule by the amino terminal peptidase, the released amino terminal inhibits collagen formation. It has been shown by immunoelectron microscopic techniques that there is retention of the amino peptide at the site of the collagen fibril.1
There are a number of subsets of antinuclear antibodies, which also have clinical predictive value: 8,140–142
• Anticentromere antibody (which is almost specific for systemic sclerosis) is particularly common in the limited cutaneous variant.143 It is usually found in patients with less severe disease and a more favorable outcome.8 Calcinosis and telangiectasia may be conspicuous, but interstitial pulmonary fibrosis is less likely.
• Scl-70 antibody (anti-DNA topoisomerase) is found in 20% to 60% of patients with systemic sclerosis, particularly the diffuse variant.144 It is also highly specific.8,145 Scl-70 antibody correlates with severe systemic involvement including pulmonary interstitial fibrosis and a poor prognosis.
• Anticentriole antibody occurs in both the limited and diffuse forms. Although these antibodies are of great diagnostic importance, they do not appear to have any pathogenetic significance.
In addition to increased quantities of collagen, the skin of early lesions of systemic sclerosis contains excessive quantities of glycosaminoglycans, notably dermatan sulfate and chondroitin 4- and 6-sulfate.134 There is some evidence to show that the increase in glycosaminoglycans may be due, at least in part, to diminished degradation; their presence is associated with water binding in vivo and presumably is therefore also responsible for edema.
Systemic sclerosis is associated with abnormalities of both humoral and cellular immunity.135 In contrast to SLE, anti-DNA antibodies are not usually present. Almost all patients, however, do possess antinuclear antibodies; these may be speckled, homogeneous, or nucleolar in type (Fig. 17.95). The last are found in 7% to 46% of patients, but are not specific, being found in a number of other connective tissue diseases.8 They form a heterogeneous group reacting against a variety of antigens, including U3-RNP (fibrillarin), RNA polymerase I, Th ribonucleoprotein, and PM-Scl, and have some prognostic significance and subtype specificity (Table 17.10).8,10 Anti-U3 RNP antibodies are present in 5% to 8% of systemic sclerosis, and are associated with African-American race, male gender, higher incidence of skeletal muscle pathology, and pulmonary arterial hypertension.136 A further study
A recent study from the Netherlands analyzing a total of 460 patients revealed interstitial lung disease to be more prevalent in localized cutaneous systemic sclerosis patients with antitopoisomerase I antibodies in comparison with patients without these antibodies (49% and 25%, respectively).146 Futhermore, localized cutaneous systemic sclerosis patients with antitopoisomerase I antibodies are more likely to progress to diffuse cutaneous systemic sclerosis.146
The presence of Ro (SS-A) and La (SS-B) antibodies suggests the coexistence of Sjögren syndrome.1 Autoantibodies against matrix metalloproteinase-3 have been detected in about 50% of patients with systemic sclerosis.147 They are significantly higher in patients with diffuse cutaneous systemic sclerosis than those with limited cutaneous systemic sclerosis, and are significantly correlated with skin fibrosis, lung fibrosis, and thickening of the renal blood vessels.147
Antibodies to types I and IV collagen have been described, but it is not clear whether they represent primary pathogenetic agents or are secondary phenomena.148 More recently, antiendothelial cell antibodies have been documented in scleroderma.149 Circulating immune complexes have been
Fig. 17.95 Systemic sclerosis: antinucleolar antibody (HEP II). By courtesy of G. Swana, MD, St Thomas’ Hospital, London, UK.
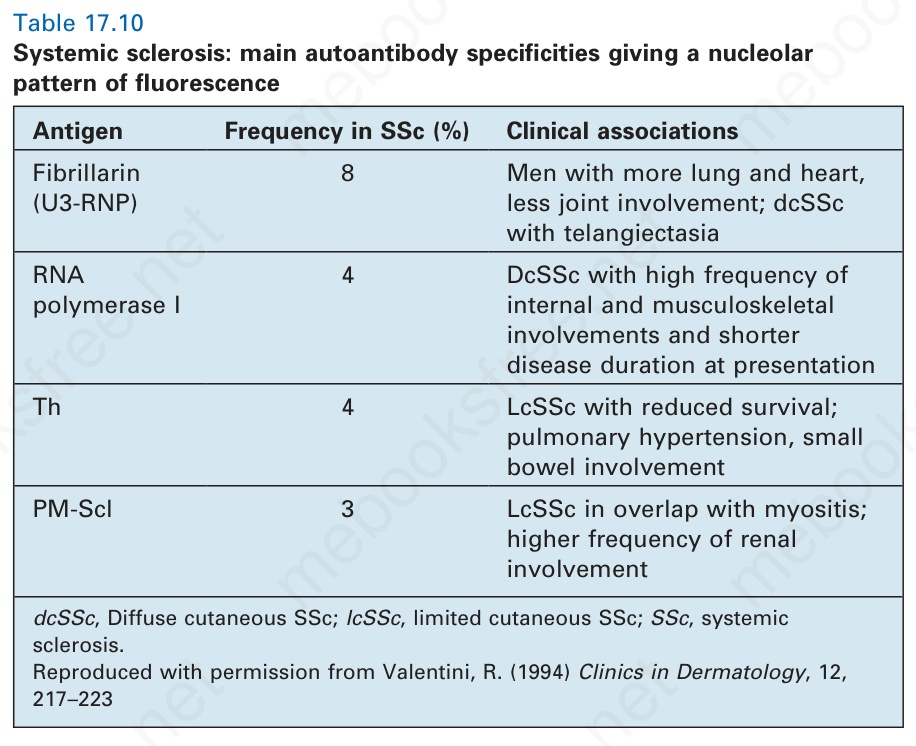
Table 17.10 Systemic sclerosis: main autoantibody specificities giving a nucleolar pattern of fluorescence
Antigen Frequency in SSc (%) Clinical associations
Fibrillarin (U3-RNP)
8 Men with more lung and heart, less joint involvement; dcSSc with telangiectasia
RNA polymerase I
4 DcSSc with high frequency of internal and musculoskeletal involvements and shorter disease duration at presentation
Th 4 LcSSc with reduced survival; pulmonary hypertension, small bowel involvement
PM-Scl 3 LcSSc in overlap with myositis; higher frequency of renal involvement
dcSSc, Diffuse cutaneous SSc; lcSSc, limited cutaneous SSc; SSc, systemic sclerosis. Reproduced with permission from Valentini, R. (1994) Clinics in Dermatology, 12, 217–223
reported, but are not a constant feature, and their significance, if any, is unknown.108,150
Various T-cell abnormalities have been reported, most of which point toward a diminished concentration of circulating T lymphocytes, particularly of the suppressor subset.103 An increased T helper:suppressor ratio has been described.151 Soluble cytotoxic T lymphocyte-associated molecule-4 (sCTLA-4) has been found to be increased in patients with diffuse cutaneous systemic sclerosis and elevated levels of sCTLA-4 appear to correlate with the diseased severity and activity.152 Clonal expansion of T cells has been detected in both blood (61%) and skin (45%) of patients with systemic sclerosis, which is significantly higher than in normal matched controls.153
805 Systemic sclerosis
Investigations have recently been directed toward the role of cytokines in the development of the fibrosis in systemic sclerosis. Evidence suggests that they are major regulators of fibroblast function and collagen synthesis. It has been proposed that in systemic sclerosis there is excessive fibroblast stimulatory activity, due, for example, to fibroblast chemotactic factors including fibronectin, collagen fragments, platelet-derived growth factor, epidermal growth factor, and C5a.1 Fibroblast growth stimulating factors, including IL-1, -2, and -3, TGF-β, and platelet-derived growth factor, are also of major importance.103,154,155
As yet no consistent strong class I or II major histocompatibility complex (MHC) antigen associations have been discovered in systemic sclerosis.10,156 There are, however, significant HLA associations with individual autoantibodies. Therefore, PM-Scl antibody correlates with HLA-DR3 and Scl-70 antibody with HLA-DR5.50,157
Susceptibility genes recently described to be associated with the development of systemic sclerosis include STAT-4, IRF5, and BANK-1.158–160
A useful working hypothesis for the pathophysiology of systemic sclerosis was suggested by Fleischmajer and Lebwohl.161 They proposed that following vascular injury, possibly caused by an autoimmune mechanism, exposure of type IV collagen or other substances leads to the recruitment of both B and T lymphocytes in addition to monocytes and mast cells. Excess T-helper cells stimulate the production of autoantibodies by B cells, whereas the activated T cells, macrophages, and mast cells secrete a variety of cytokines, which in turn promote collagenosis.
Histologically, the edema of the early stage produces a picture that is indistinguishable from scleredema of Buschke.162,163 In an established lesion the epidermis sometimes appears normal or there may be loss of the rete ridge pattern. There is often increased pigmentation of the basal cells, and melanophages are common in the superficial dermis. The characteristic change is that of thickening of the dermis by broad, elongated, swollen collagen bundles that often appear orientated parallel to the surface epithelium (Figs 17.96 and 17.97). The individual fiber borders are frequently indistinct, giving the collagen a rather homogenous appearance. The elastic fibers are usually unaffected. The fibrosis characteristically involves the subcutis and therefore fat cells are usually incorporated into the dermis.164 Atrophic skin appendages, particularly eccrine sweat glands, are a common feature.
The arteries, especially the digital vessels, typically show endothelial cell swelling, intimal thickening, and medial hypertrophy. Later they may become hyalinized (Fig. 17.98). In early lesions, endothelium-associated platelets are significantly increased in number.108 Fibrin deposition is sometimes present and occasionally complete occlusion results in digital ulceration and gangrene. With chronicity there is a progressive reduction in the number of vessels, particularly in the more superficial dermis.108 Perineural fibrosis is sometimes a feature and calcification is not uncommon (Figs 17.99 and 17.100).
In early lesions there may be a chronic inflammatory cell infiltrate comprising lymphocytes, histiocytes, and a few plasma cells around blood vessels and at the interface between the dermis and the subcutaneous fat.1,165 T-helper cells predominate and increased numbers of dermal Langerhans cells have been described.108 Mast cells, usually activated, are often present in increased numbers.166 Palisaded neutrophilic and granulomatous dermatitis has been reported in a single patient with limited systemic sclerosis.167 Deposition of amyloid in the dermis and subcutis in a patient with CREST syndrome has also been reported.168
It is usually not possible to distinguish localized scleroderma (morphea) from systemic sclerosis on histologic grounds, although the epidermis is
806 Idiopathic connective tissue disorders
usually normal in the localized form and vascular changes are less severe. In contrast, the inflammatory cell infiltrate is often heavier in the localized variant and commonly affects the reticular dermis.165
Examination of skeletal muscle may reveal focal scarring and a chronic inflammatory cell infiltrate (Figs 17.101 and 17.102).169 Features of muscle degeneration (vacuolation, homogenization with eosinophilia, and loss of cross-striations) and regeneration (basophilia and sarcolemmal nuclear proliferation) similar to that seen in dermatomyositis may also be present. In the sclerotic phase there is atrophy and fibrosis.
Macroscopic examination of the kidney often reveals multiple infarcts, foci of hemorrhage, and occasionally, the features of renal cortical necrosis.68 The histologic appearances are similar to those of malignant hypertension
and are characterized by the presence of fibrinoid necrosis, which particularly affects the interlobular and arcuate arteries, and ischemic glomerulosclerosis; an inflammatory cell infiltrate is not a feature.63,170 Characteristic of systemic sclerosis is the presence of edema and mucoid change in the initima of the interlobular arteries. There is also increased cellularity, giving rise to a characteristic ‘onion skin’ appearance with reduction in the lumen of the vessel.
The histologic features of sclerodermatous interstitial pneumonitis are indistinguishable from those seen in idiopathic fibrosing alveolitis. Early stages are characterized by intra-alveolar edema with reactive pneumocytes and a variable infiltrate of macrophages, lymphocytes, and occasional neutrophils.50,171 Interstitial accumulations of lymphocytes and plasma cells are evident, sometimes associated with focal lymphoid hyperplasia; in older lesions, these are accompanied by the deposition of glycosaminoglycan-rich new collagen. End-stage disease is characterized by the development of variably sized cysts lined by metaplastic bronchiolar epithelium and containing abundant collagen and hyperplastic smooth muscle in their walls.172
807 Localized scleroderma (morphea)
has found mild internal involvement consisting of abnormal lower sphincter pressure and peristaltic failure in the esophagus and slightly impaired carbon monoxide diffusion in the lung in up to 19% of patients.18 These abnormalities do not result in clinical symptoms and do not affect prognosis adversely. A rare case has been documented in which morphea induced severe extrapulmonary thoracic restriction.19 Extracutaneous involvement is present in about one-fifth of the children, and includes articular, neurological, vascular, ocular, gastrointestinal, respiratory, cardiac, and renal manifestations, in decreasing order of frequency.16 Although the plaques and, to a lesser extent, the linear lesions often improve with time, the contractures and hemiatrophy are permanent.3 Imaging studies frequently reveal muscle atrophy and leg length discrepancy.20 Localized scleroderma may occur after trauma, laparoscopy, radiotherapy, tattooing, and silicone implants.21–27 It has also been described in association with bromocriptine, balicatib, valproic acid, and ibuprofen therapy.28–31 Localized scleroderma has also been reported following silica dust exposure.32
The features of pulmonary hypertension are commonly present, particularly in patients with the CREST variant. Muscular arterioles are predominantly affected, although in late stages venules may also be involved, and show medial muscular hypertrophy and concentric myxoid-rich new collagen deposition in the intima with variable reduction in the diameter of the lumen.172 In late stages, muscular atrophy and medial elastosis may be evident. Focal lymphocytic/plasma cell endovasculitis has been documented, suggesting a possible autoimmune pathogenesis.172 Fibrosis of pulmonary veins and venules can be similar to the changes observed in pulmonary veno-occlusive disease.173 Pulmonary hypertension correlates with the presence of anticentromere antibody. Bronchiolitis predominantly affects the terminal and respiratory bronchioles. In addition to chronic inflammation, bronchiolar squamous metaplasia and variable scarring with luminal constriction may be seen.172
A recent study involving in total of 344 patients with pediatric-onset and adult-onset localized scleroderma detected disease recurrence in 27% and 17% of the patients, respectively.33 Recurrences were more frequent in patients with linear localized scleroderma occurring on the limbs and were independent of age at disease onset.33
The precise relationship between localized scleroderma and systemic sclerosis is uncertain. Because of clinical and pathological overlap, some authors believe that the two conditions represent extreme ends of a spectrum of connective tissue damage in a manner similar to the relationship between discoid and systemic lupus erythematosus. Indeed, patients rarely have both morphea and progressive systemic sclerosis (the former usually preceding the latter); this phenomenon occurs so infrequently, however, that most believe that the relationship is purely coincidental.2 Alternatively, the features of these two disorders may merely represent a common manifestation of tissue damage caused by quite different mechanisms, analogous to the wide range of pathogenetic factors which may result in the histologic appearance of allergic vasculitis.
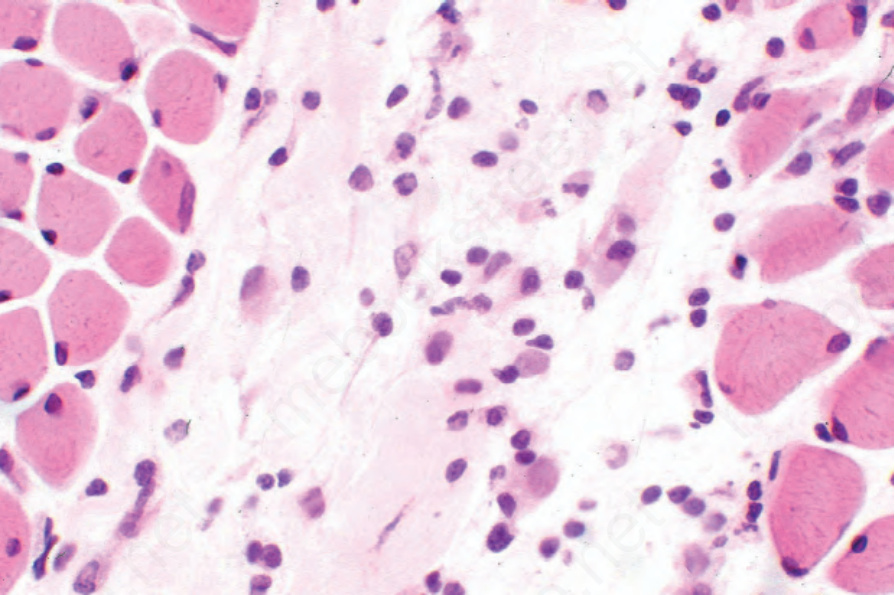
Fig. 17.101 Systemic sclerosis: myositis characterized by a lymphohistiocytic infiltrate and focal skeletal muscle regeneration.
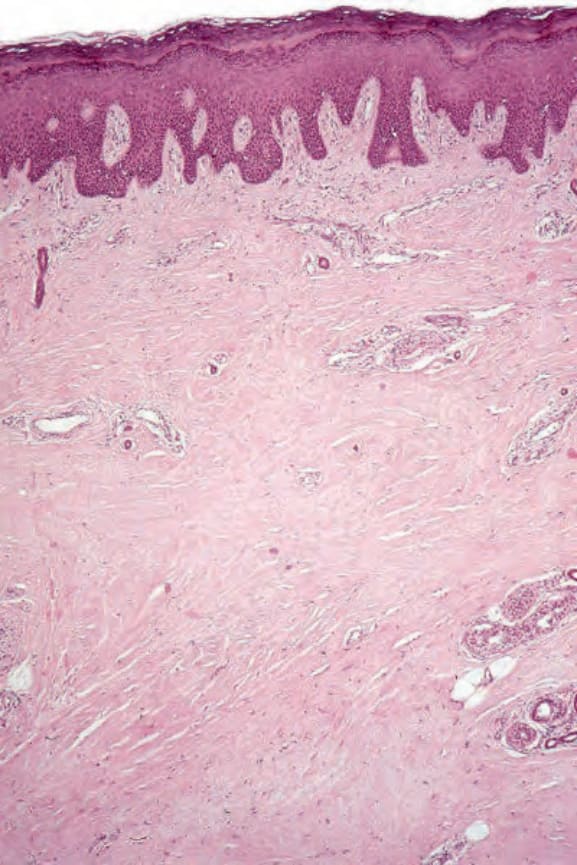
Fig. 17.96 Systemic sclerosis: scanning view of acral skin showing dermal fibrosis. The specimen was a foot amputation performed because of severe vascular involvement.
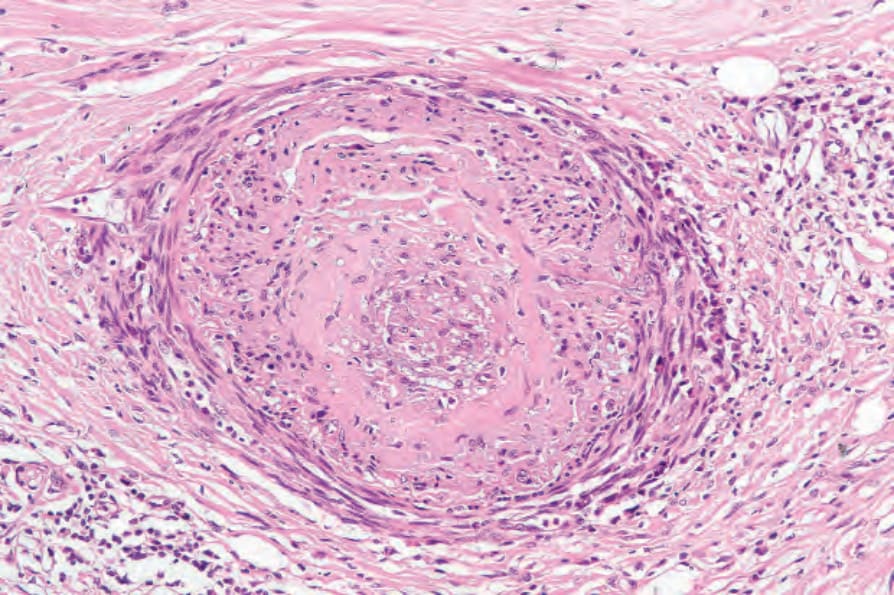
Fig. 17.98 Systemic sclerosis: severe vascular involvement characterized by intimal fibrosis and obliteration of the lumen. Note the surrounding chronic inflammation and scarring.
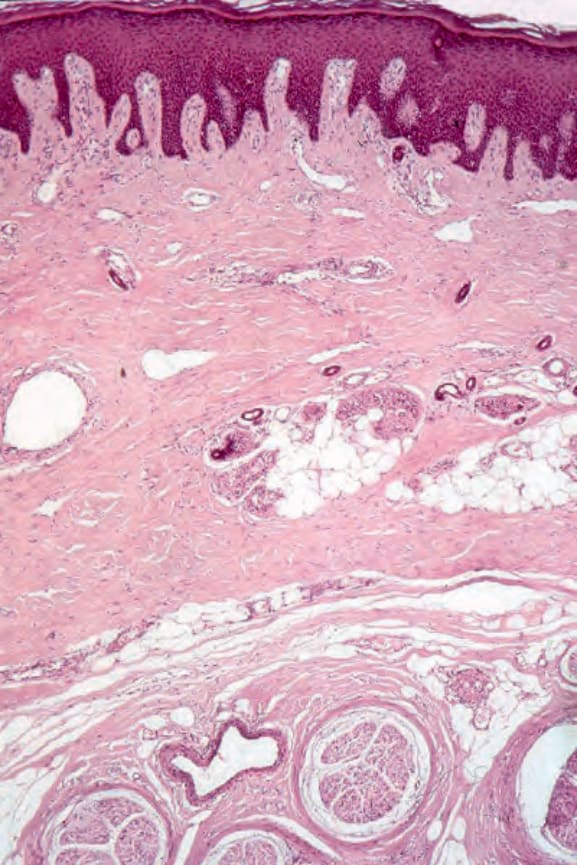
Fig. 17.99 Systemic sclerosis: there is dramatic perineural fibrosis.
The most important gastrointestinal lesion is atrophy with fibrosis of the esophageal smooth muscle; similar changes may also develop in the small and large intestines. Vascular myointimal proliferation with luminal narrowing is also usually evident.70 Reflux esophagitis may show erosions and areas of ulceration in addition to chronic inflammatory changes. In contrast to conventional diverticulae, those of systemic sclerosis are composed of all layers of the bowel wall.
Clinical features Localized scleroderma includes a variety of conditions, which may arise independently, but which frequently occur together:
• plaque-form (the most common variant),
• bullous morphea,
• guttate lesions,
• linear morphea including facial hemiatrophy,
• generalized morphea,
• subcutaneous scleroderma (morphea profunda),
• disabling pansclerotic morphea of children.2,34–36
Early myocardial changes are characterized by necrosis of muscle fibers accompanied by a chronic inflammatory cell and histiocytic infiltrate.62 Subsequent fibrosis affects the right and left ventricles with equal frequency.61 The major coronary arteries appear normal in systemic sclerosis, but arteriolar, endothelial, and intimal proliferation accompanied by mural scarring is common.174
In active lesions, synovial biopsies show a heavy surface fibrin deposit.74 There is adjacent chronic synovitis with an admixture of lymphocytes and plasma cells. Lymphoid follicles with germinal center formation as seen in rheumatoid arthritis are not a feature. With chronicity, synovial scarring supervenes.
Plaque-form and linear morphea Plaque-form and linear morphea are more common in females (3 : 1) and, in contrast to progressive systemic sclerosis, often occur in childhood.1 Linear morphea develops before the end of the first decade in up to 20% of patients and by the fourth decade in up to 75%. Localized plaques occur a little later in life, although 75% of patients are between 20 and 50 years of age at presentation.