疾病定義與分類
系統性硬化症 (systemic sclerosis)
臨床特徵 (Clinical Features)
進行性系統性硬化症 (progressive systemic sclerosis) 包含兩個主要變異型:
-
在較嚴重的瀰漫型 (diffuse form) 中,病人除了內臟(尤其是腎臟、肺臟、心臟、食道與腸道)受侵犯外,還在掌指 (metacarpophalangeal) 與蹠趾關節 (metatarsophalangeal joints) 近端出現廣泛的皮膚病灶(近端硬皮病,proximal scleroderma)。此病常以急性發作起病,伴隨疲倦、體重減輕、關節痛 (arthralgia) 與腕隧道症候群 (carpal tunnel syndrome)。肌腱摩擦音 (tendon friction rubs) 是其特徵。常可見 anti-Scl-70 (anti-DNA topoisomerase) 與 RNA polymerase III 抗體,預後一般不佳。
-
另一個主要變異型則與局限性周邊皮膚硬化 (limited peripheral cutaneous sclerosis) 相關,除食道侵犯、小腸吸收不良 (small intestinal malabsorption) 與肺高壓 (pulmonary hypertension) 外,並無嚴重的全身性疾病;其預後通常較佳。此型與抗著絲點抗體 (anticentromere antibody) 相關。系統性硬化症偶爾也會在沒有皮膚表現的情況下被記錄到(無硬皮病變異型,sine scleroderma variant),亦曾描述重疊症候群 (overlap syndromes)。局限性硬皮病也包含 CREST(calcinosis、Raynaud phenomenon、esophageal dysfunction、sclerodactyly、telangiectasis)症候群(Thibierge-Weissenbach syndrome,肢端硬化症 acrosclerosis),其皮膚疾病表現局限於手指與腳趾(指趾硬化,sclerodactyly)及顏面(見下文)。其他全身型變異型包括硬皮病肌炎 (sclerodermatomyositis)、混合性結締組織病 (MCTD) 以及化學物質誘發的硬皮病樣症候群 (chemically induced scleroderma-like syndromes)。
由於系統性硬化症可侵犯多種系統,病人可能主要由皮膚科、風濕科、腎臟科或其他專科醫師照護,導致難以確定此病的確切發生率;估計約為每年每百萬人口 20 例新發個案。在一個大型病例系列中,瀰漫型與局限型一樣常見。約有百分之十被歸類為重疊症候群。此病在家族中較常發生,被視為此病已確認的最強危險因子。然而,每位家族成員的絕對風險很低。系統性硬化症有明顯的女性優勢(3–4 : 1);雖然任何年齡層都可能受影響,但病人最常於第四、第五與第六個十年發病。年輕的黑人女性構成一個風險特別高的明確亞群。偶有家族性病例被記錄到,通常見於兒童。幼年型系統性硬化症 (juvenile systemic sclerosis) 約佔系統性硬化症病人的百分之三。在 8 歲之前並無性別偏好。與成人發病的系統性硬化症相比,幼年型系統性硬化症伴隨較高的重疊症候群發生率(尤其與多發性肌炎/皮肌炎 polymyositis/dermatomyositis 重疊)、血清中不同組合的抗體(anti-PM-Scl 與 anti-U1-RNP),以及較佳的存活率。心肺疾病對幼年型系統性硬化症的存活具有預測價值。
由於缺血,指尖出現指腹萎縮 (pulp atrophy) 與末節指骨吸收 (absorption of the terminal phalanges),指尖常無法突出於指甲游離緣之外(圖 17.83–17.85)。指甲可能出現縱向脊紋與脆弱,甚至可能脫落。受侵犯的皮膚具有非常特殊的外觀,呈現光亮、平滑且相當蠟樣。病人的手腳活動度常明顯減低,屈曲攣縮 (flexion contractures) 很常見(圖 17.86)。在進展期,許多病人會出現戲劇性的僵硬、缺乏表情的臉孔,伴隨鳥喙鼻 (beaked nose)、變薄的嘴唇、口周溝紋與皺褶,以及無法完全張口(圖 17.87 與 17.88)。也可能注意到下眼瞼緊繃,前額可能顯得平滑而無皺紋。潰瘍是常見的併發症,尤其在緊繃的皮膚被拉伸覆蓋於易受外傷的骨突處時(圖 17.89)。
系統性硬化症的整體死亡率高,5 年存活率從 34% 到 73% 不等。已證實皮膚增厚的改善與存活改善相關。年齡較大的病人與男性通常預後最差。
局限性皮膚系統性硬化症 (Limited cutaneous systemic sclerosis) 在局限型變異型中,皮膚表現常先影響雙手,包括早期水腫、硬化以及晚期萎縮等階段。水腫的特徵為非凹陷性 (nonpitting)、雙側且對稱。手指常被描述為呈香腸狀外觀 (sausage-like appearance)(圖 17.82)。顏面、前臂、足部與腿部有時也會受影響。隨著水腫消退,皮膚變得增厚緊繃並固著於皮下組織。典型上,手指變得逐漸尖細,並且
血管變化很常見,包括周邊壞疽 (peripheral gangrene)、手指自截 (digital autoamputation) 與雷諾現象 (Raynaud phenomenon)。後者出現得如此頻繁(在局限型與瀰漫型中皆然),以致常被教導:有雷諾現象的病人,在未證實之前都應假定患有系統性硬化症。在局限性皮膚系統性硬化症中,雷諾現象可能比皮膚病灶的發作早出現許多年,這在很大比例
的病人中發生。系統性硬化症一個有用的診斷特徵是許多甲褶微血管 (nail fold capillaries) 的喪失與其餘微血管的擴張。近期已證實甲褶微血管鏡 (nail fold capillaroscopy) 的異常與系統性硬化症的瀰漫型、皮膚侵犯的嚴重度、受影響皮節 (tracts) 的數目,以及 anti-Scl-70 抗體的存在相關。
CREST 症候群的皮膚變化主要位於掌指關節遠端,雖然手背與口部有時也會受影響。CREST 症候群中的發炎期是持續性的。
雷諾現象,無論是單獨出現或合併腫脹臃腫的手指,是目前為止最常見的表現方式,且毛細血管擴張 (telangiectases) 往往比瀰漫型系統性硬化症的病人多得多(常多達數百個)。它們特別影響手指與手部、
顏面、舌頭與黏膜(圖 17.90–17.92)。此種硬皮病變異型所見的毛細血管擴張可能難以與遺傳性出血性毛細血管擴張症 (hereditary hemorrhagic telangiectasia) 所見者區分。食道功能障礙與瀰漫型變異型所見者相同,但傾向於更嚴重並影響大多數病人。
並在局限性皮膚系統性硬化症變異型中較常見。可能有超過一種相關的自體免疫疾病與 CREST 症候群並存。有人認為合併原發性膽汁性肝硬化 (primary biliary cirrhosis) 的病人構成此病的一個特殊亞群,傾向於較輕微且預後較佳。曾報告一例罕見的、抗粒線體抗體 (antimitochondrial antibody) 陽性的原發性膽汁性肝硬化於急性心肌梗塞後發生。雖然臨床上具意義的原發性膽汁性肝硬化僅在 2.5% 的系統性硬化症病人中發生,近期一項研究已證實在 15% 的系統性硬化症病人中存在抗粒線體抗體。系統性硬化症中也發現原發性膽汁性肝硬化與自體免疫性肝炎 (autoimmune hepatitis) 之間的重疊症候群。一項多中心的法國–義大利研究發現,21% 的病人同時存在系統性硬化症與自體免疫疾病,其中以 Sjögren 症候群(12%)與甲狀腺炎(6%)為最常見的相關。伴隨自體免疫疾病的系統性硬化症病人似乎呈現較輕微的病程。系統性硬化症在約 5% 的病人中與類風濕性關節炎 (rheumatoid arthritis) 相關,可能代表一個與 HLA-DR3、HLA-DR7、HLA-DR11 與 HLA-DRw53 相關的特殊疾病亞群。在一個超過 2000 名系統性硬化症病人的大型世代中,1.6% 的病人確立與 ANCA 陽性血管炎 (ANCA-positive vasculitis) 的相關。一個重疊
「CREST 症候群」此一名稱的價值,自從發現許多局限性皮膚疾病的病人並未符合其全部標準,且觀察到「CREST」表現可能見於許多瀰漫型疾病的病人之後,已大幅降低。例如,有一種變異型由手指壞死、雷諾現象與抗著絲點抗體組成,而無指趾硬化。此名詞可能應被廢棄,並將所有局限性遠端疾病的病人歸類為單一亞型。然而,考量 CREST 症候群在當前文獻中出現的頻率,至少在可預見的未來,這不太可能發生。原本認為局限型變異型與相對良性的結果相關,但現已知若追蹤病人足夠長的時間,相當比例的病人會發展出嚴重的肺高壓及其後遺症。也有增加的風險發展出 Sjögren 症候群、膽汁性肝硬化、盤狀紅斑性狼瘡 (discoid lupus erythematosus),以及甲狀腺功能障礙,如 Hashimoto 甲狀腺炎與 Graves 病。在 14% 的系統性硬化症病人中發現 Sjögren 症候群
系統性硬化症與冷凝球蛋白血症性血管炎 (cryoglobulinemic vasculitis) 之間的重疊在 1.6% 的病人中被偵測到,通常存在 C 型肝炎病毒 (hepatitis C virus) 感染。
色素沉著過度 (Hyperpigmentation),從淺棕色到深古銅色,令人聯想到 Addison 病,是一種常見的表現,且常伴隨小灶的色素脫失 (hypopigmentation),形成特徵性的「鹽與胡椒 (salt and pepper)」外觀。色素變化特別影響手背與前臂,以及上胸部與背部。有時伴隨的色素脫失程度如此明顯,以致類似白斑 (vitiligo)。在例外情況下,於缺乏相關硬化的情況下出現的皮膚色素異常,可代表早期系統性硬化症的初始表現。
系統性硬化症一種罕見的皮膚表現,特徵為多發且不可移動的小丘疹或結節,使病灶呈現所謂的鵝卵石樣外觀 (cobblestone appearance)。一般認為此種表現型態是由於纖維化過程阻塞淋巴管道所造成的淋巴管擴張 (lymphangiectasia) 的結果。
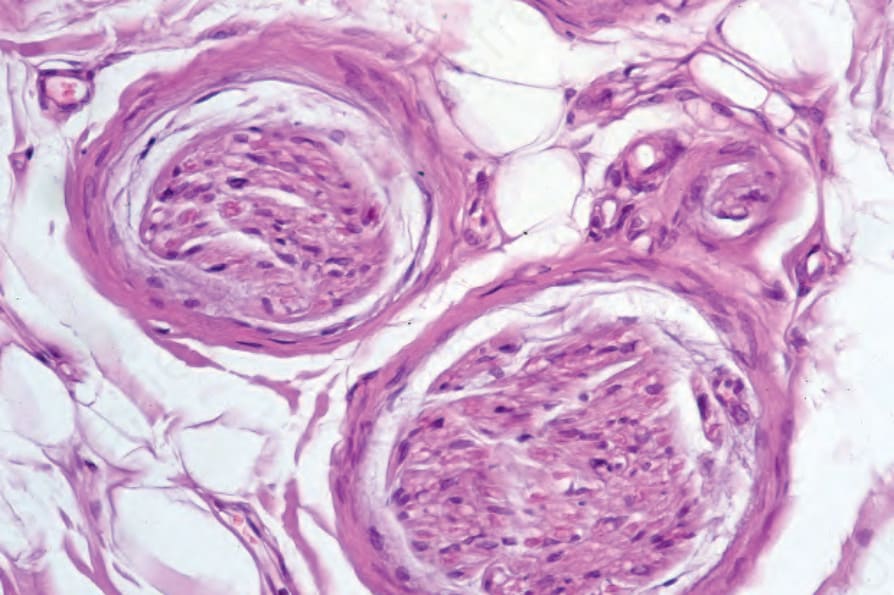
圖 17.100:系統性硬化症 (systemic sclerosis):高倍視野。
Fig. 17.100 Systemic sclerosis: high-power view.
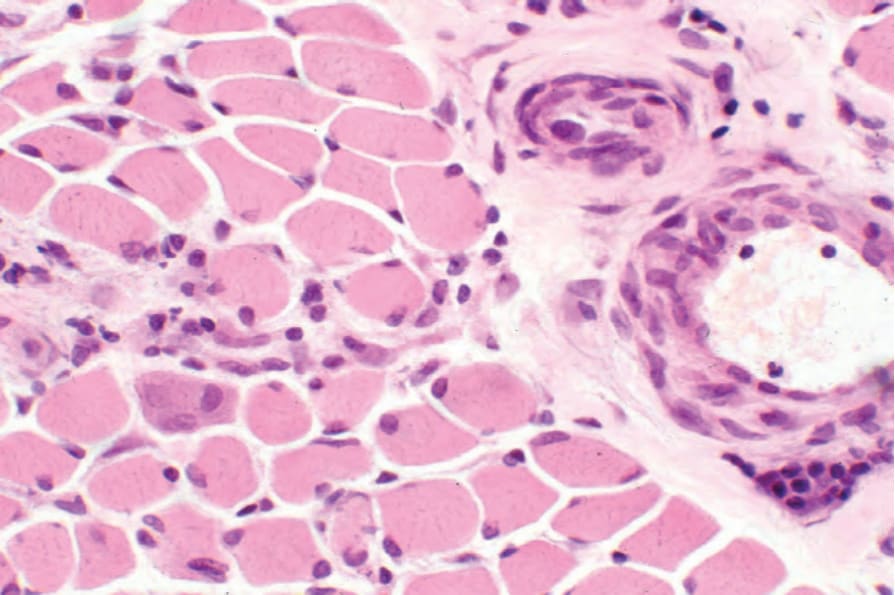
圖 17.102:系統性硬化症 (systemic sclerosis):注意視野左側的局灶性細胞質嗜鹼性 (focal cytoplasmic basophilia)。
Fig. 17.102 Systemic sclerosis: Note the focal cytoplasmic basophilia on the left side of the field.
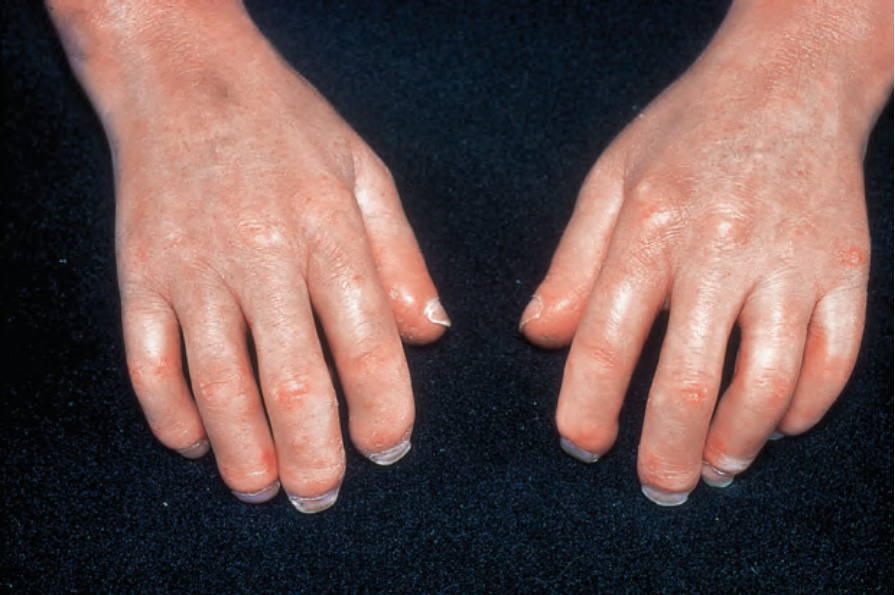
圖 17.82:系統性硬化症 (systemic sclerosis):早期階段,顯示特徵性的腫脹、香腸狀手指。承蒙 the Institute of Dermatology, London, UK 提供。
Fig. 17.82 Systemic sclerosis: early stage showing characteristic swollen, sausage-shaped fingers. By courtesy of the Institute of Dermatology, London, UK.
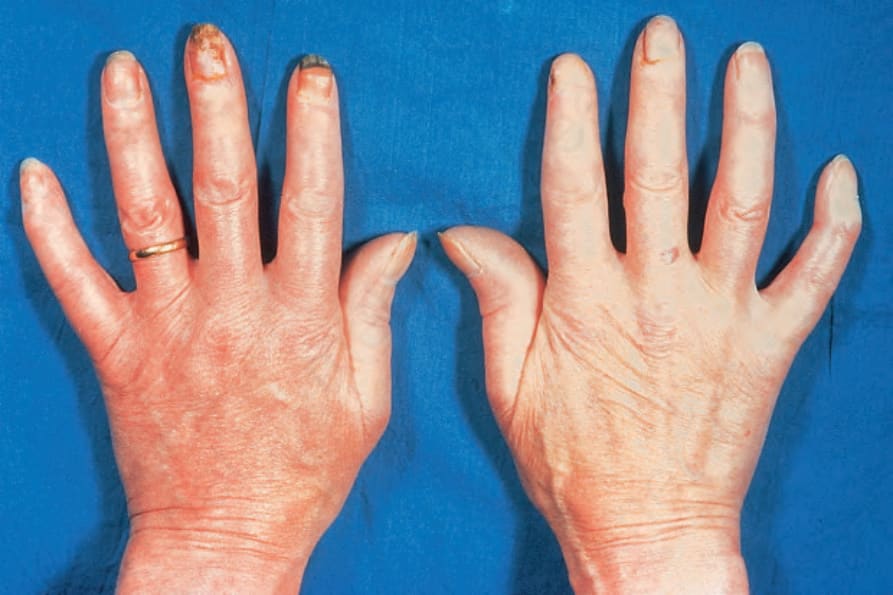
圖 17.83:系統性硬化症 (systemic sclerosis):手指呈紅斑且光亮,皮膚顯得略微固著。承蒙 R.A. Marsden, MD, St George’s Hospital, London, UK 提供。
Fig. 17.83 Systemic sclerosis: the fingers are erythematous and shiny and the skin appears slightly bound down. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
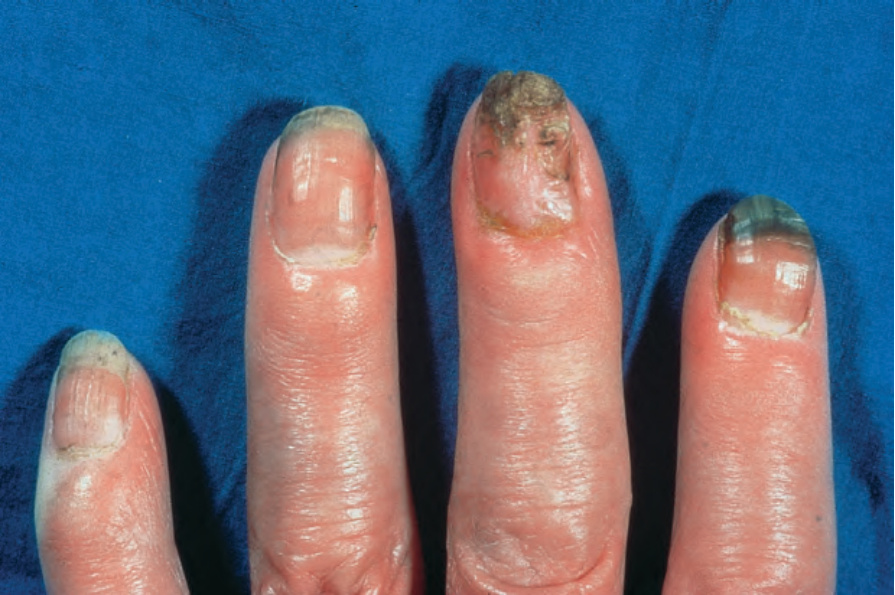
圖 17.84:系統性硬化症 (systemic sclerosis):指尖呈尖細狀。承蒙 R.A. Marsden, MD, St George’s Hospital, London, UK 提供。
Fig. 17.84 Systemic sclerosis: the fingertips are tapered. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
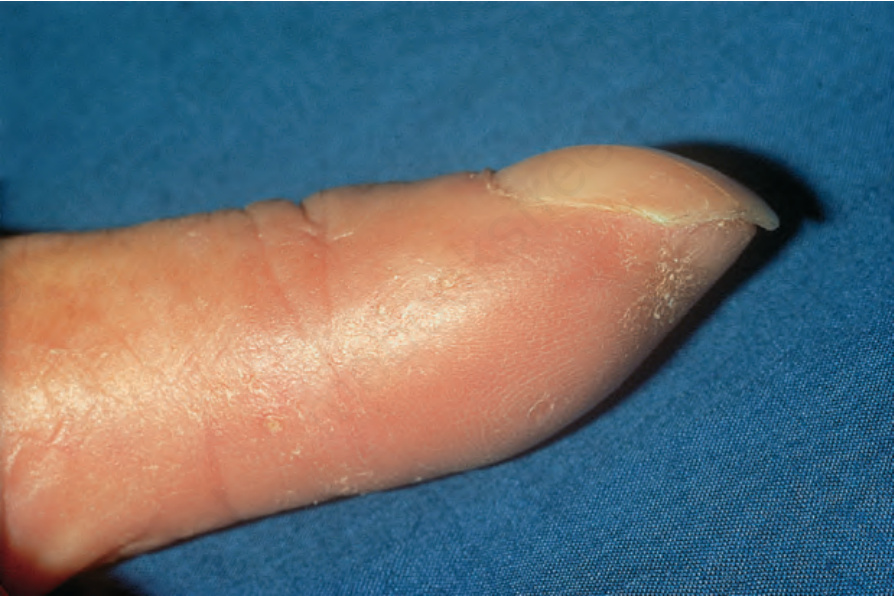
圖 17.85:系統性硬化症 (systemic sclerosis):注意指尖明顯的萎縮。承蒙 R.A. Marsden, MD, St George’s Hospital, London, UK 提供。
Fig. 17.85 Systemic sclerosis: note the marked atrophy of the fingertip. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
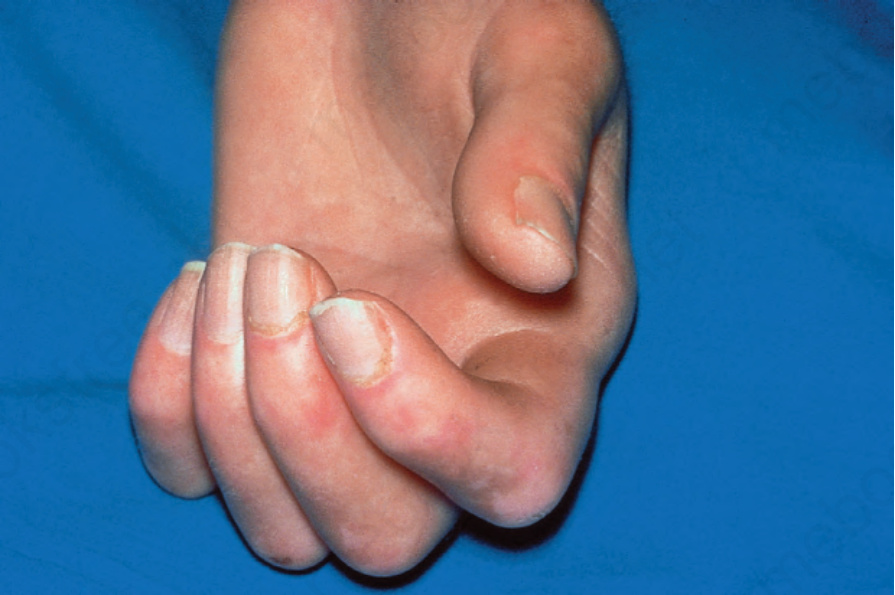
圖 17.86:系統性硬化症 (systemic sclerosis):注意屈曲攣縮 (flexion contractures)。皮膚固著且顯得萎縮。有甲周紅斑 (periungual erythema)。承蒙 R.A. Marsden, MD, St George’s Hospital, London, UK 提供。
Fig. 17.86 Systemic sclerosis: note the flexion contractures. The skin is bound down and appears atrophic. There is periungual erythema. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
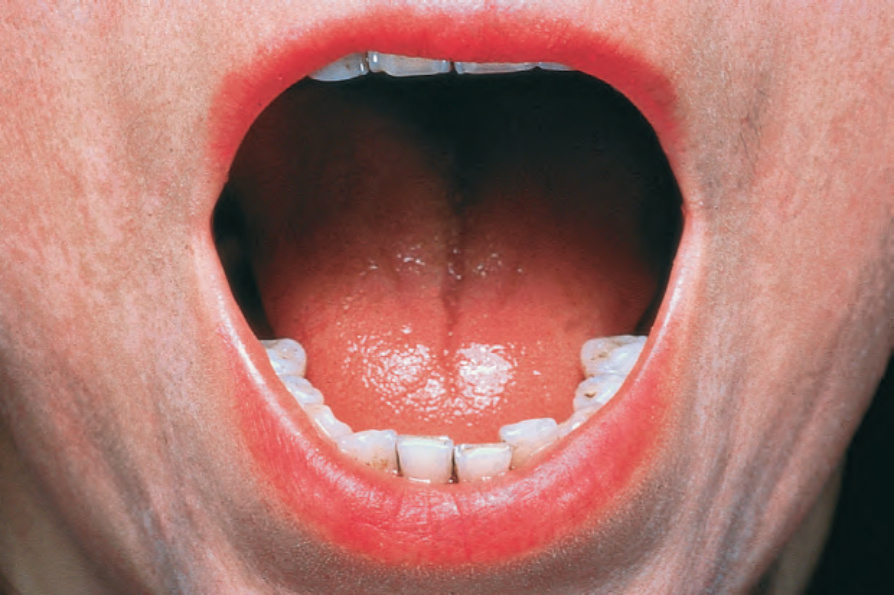
圖 17.87:系統性硬化症 (systemic sclerosis):有口周瘢痕 (perioral scarring) 伴萎縮。承蒙 the Institute of Dermatology, London, UK 提供。
Fig. 17.87 Systemic sclerosis: there is perioral scarring with atrophy. By courtesy of the Institute of Dermatology, London, UK.
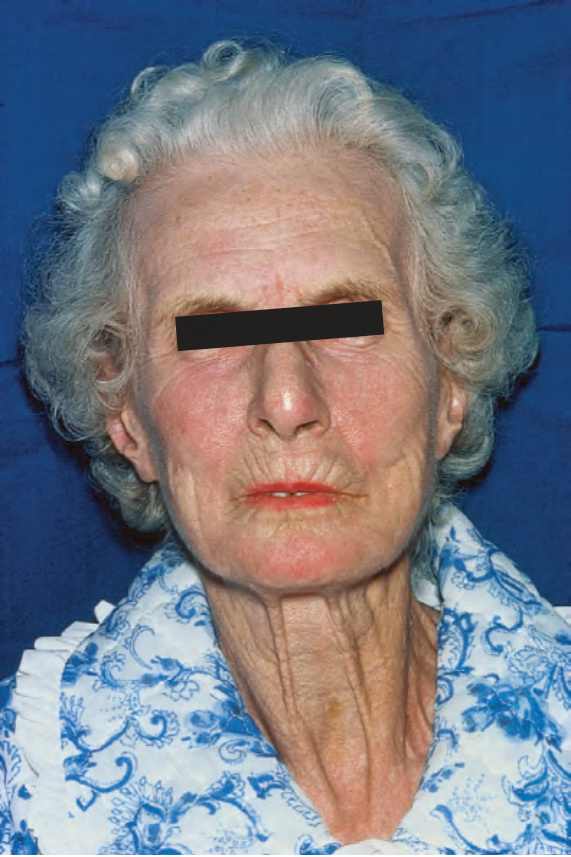
圖 17.88:系統性硬化症 (systemic sclerosis):注意變薄的嘴唇與特徵性的放射狀溝紋。承蒙 R.A. Marsden, MD, St George’s Hospital, London, UK 提供。
Fig. 17.88 Systemic sclerosis: note the thinned lips and characteristic radiating furrows. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
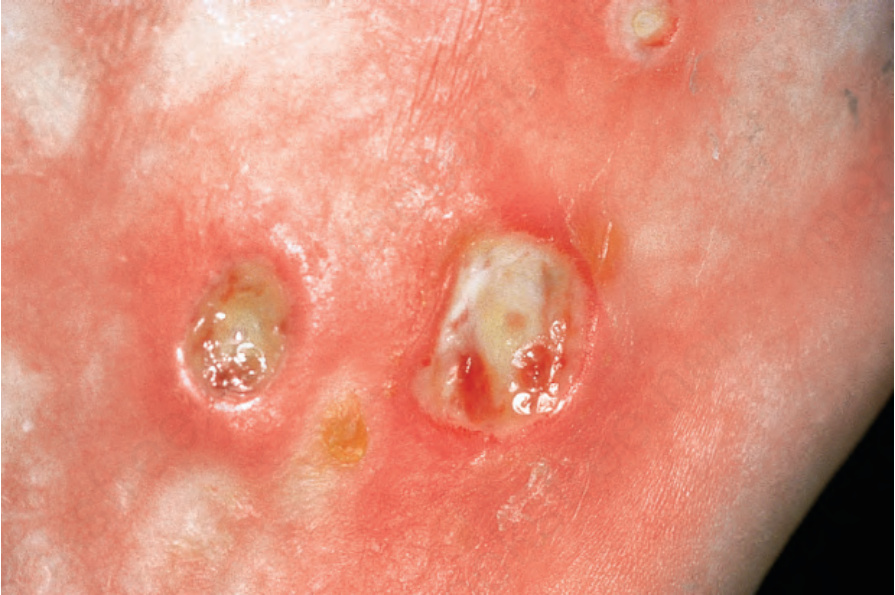
圖 17.89:系統性硬化症 (systemic sclerosis):潰瘍是一種特別令人痛苦的併發症。承蒙 the Institute of Dermatology, London, UK 提供。
Fig. 17.89 Systemic sclerosis: ulceration is a particularly distressing complication. By courtesy of the Institute of Dermatology, London, UK.
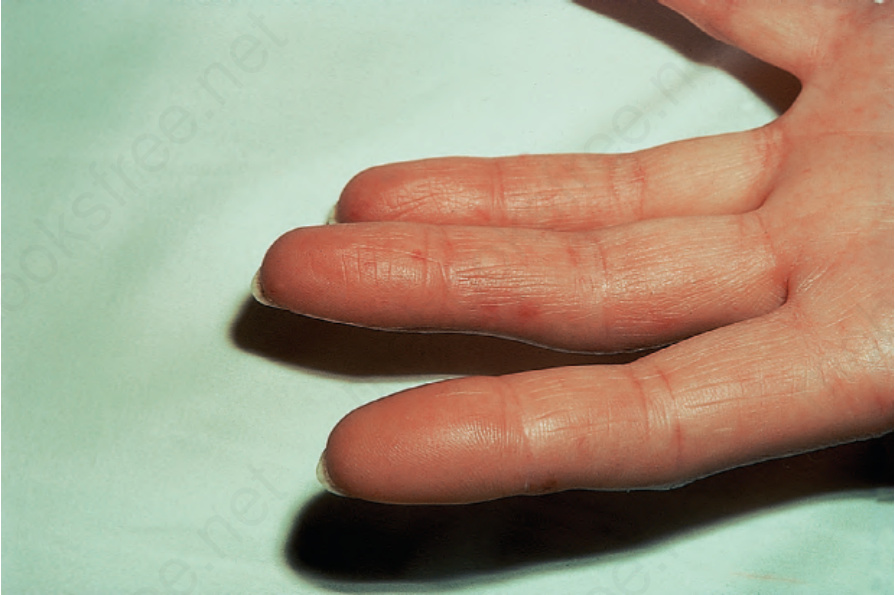
圖 17.90:系統性硬化症 (systemic sclerosis):如這些手指上所見的毛細血管擴張 (telangiectasia) 是常見的發現。承蒙 S. Parker, MD, West Middlesex Hospital, London, UK 提供。
Fig. 17.90 Systemic sclerosis: telangiectasia as seen on these fingers is a common finding. By courtesy of S. Parker, MD, West Middlesex Hospital, London, UK.
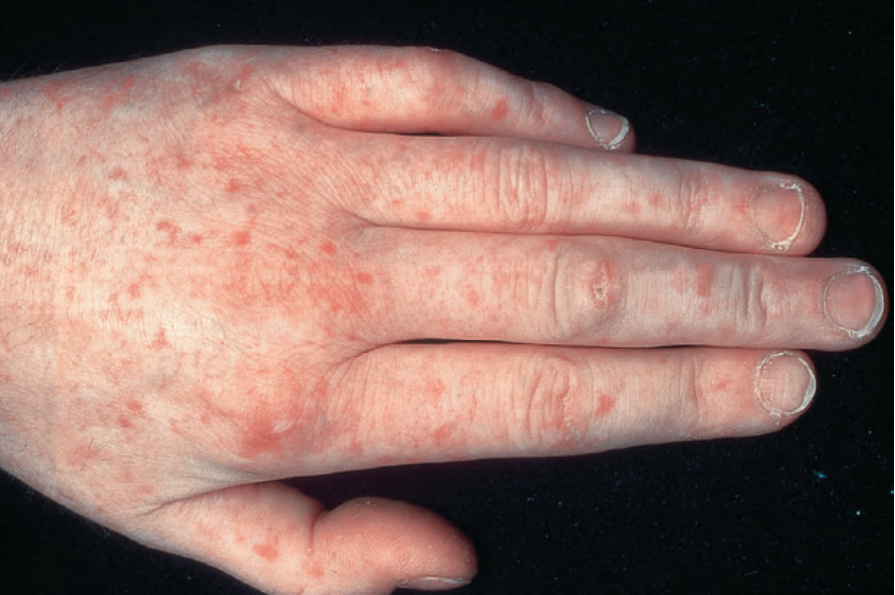
圖 17.91:系統性硬化症 (systemic sclerosis):存在眾多毛細血管擴張 (telangiectases)。手部是常受影響的部位。承蒙 the Institute of Dermatology, London, UK 提供。
Fig. 17.91 Systemic sclerosis: numerous telangiectases are present. The hand is a commonly affected site. By courtesy of the Institute of Dermatology, London, UK.
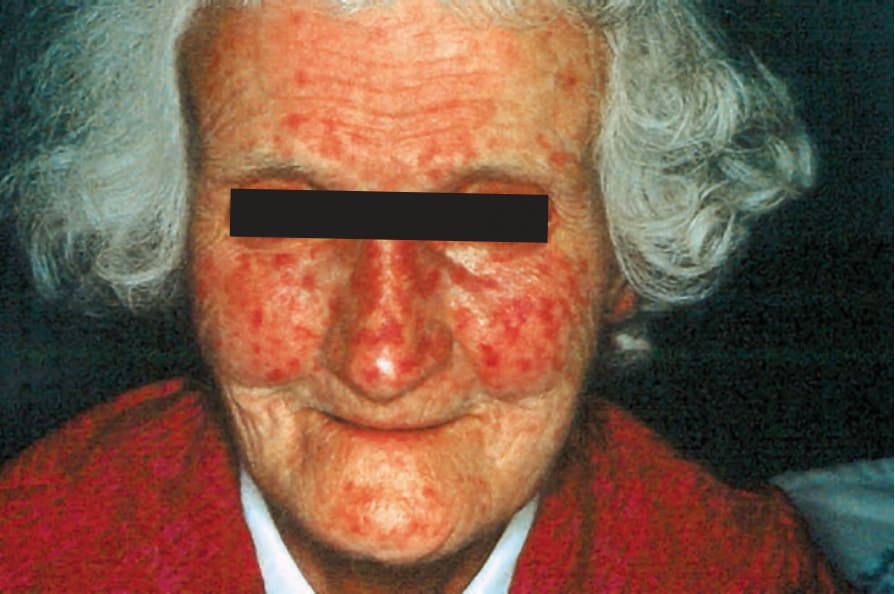
圖 17.92:系統性硬化症 (systemic sclerosis):如此病人所見的廣泛毛細血管擴張 (telangiectasia),較常是局限型變異型的特徵。承蒙 S. Parker, MD, West Middlesex Hospital, London, UK 提供。
Fig. 17.92 Systemic sclerosis: extensive telangiectasia as seen in this patient is more often a feature of the limited variant. By courtesy of S. Parker, MD, West Middlesex Hospital, London, UK.
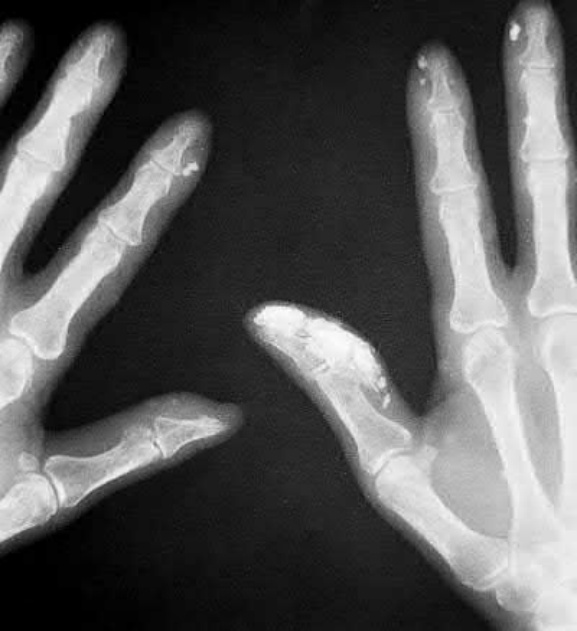
圖 17.94:系統性硬化症 (systemic sclerosis):X 光片顯示一個更廣泛的例子。承蒙 R.A. Marsden, MD, St George’s Hospital, London, UK 提供。
Fig. 17.94 Systemic sclerosis: radiograph demonstrating a more widespread example. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
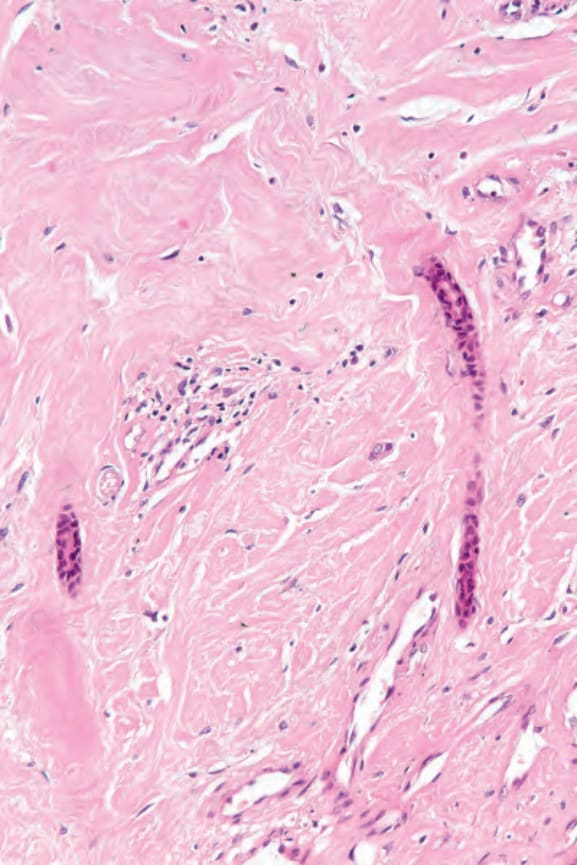
圖 17.97:系統性硬化症 (systemic sclerosis):真皮呈均質化 (homogenized)。注意受壓迫的外分泌汗腺導管 (eccrine ducts)。
Fig. 17.97 Systemic sclerosis: the dermis is homogenized. Note the compressed eccrine ducts.
對系統性硬化症病人的早期檢查應建立呼吸、心臟與腎臟功能的基準值,以便能準確監測病程進展。
肺臟的臨床侵犯很常見,是發病率與死亡率的重要原因。一般認為大多數病人都會出現程度不一的肺臟侵犯。有三種主要型態:間質性肺炎 (interstitial pneumonitis)、細支氣管炎 (bronchiolitis) 與肺血管疾病 (pulmonary vascular disease)。
-
肺間質侵犯導致運動時呼吸短促與非生產性咳嗽 (nonproductive cough)。呼吸困難 (dyspnea) 是將近 60% 未經篩選的瀰漫型硬皮病病人的特徵。症狀在合併 Sjögren 症候群的病人中傾向於特別明顯。間質性肺病是系統性硬化症最常見的死因。有證據顯示具 HLA-DR3/-DRw52A 單倍型與 anti-Scl-70 抗體的病人,肺纖維化 (pulmonary fibrosis) 的風險增加。
-
細支氣管炎在大約 13% 到 25% 的病人中明顯,但通常無症狀。
-
肺高壓 (pulmonary hypertension),可能是系統性硬化症的原發表現,或繼發於間質纖維化而發展,在局限型疾病的病人中較常見。一項研究發現,停經後狀態(無論是否存在 HLA-B35)是發展出肺高壓的主要危險因子。
皮膚變化常伴隨皮膚鈣質沉著症 (calcinosis cutis) 的發展,尤其在女性。其型態為失養性 (dystrophic),由於羥磷灰石 (hydroxyapatite) 結晶沉積所致。病人沒有鈣與磷代謝的異常,其血清鹼性磷酸酶 (alkaline phosphatase) 濃度正常。鈣質沉積的部位特別包括拇指的掌指關節與指尖,雖然前臂伸側、臀部、鷹嘴滑囊 (olecranon bursae) 與髕前區 (prepatellar region) 也可能受影響(圖 17.93 與 17.94)。這些沉積物常極為疼痛,且常伴隨潰瘍與白色顆粒狀鈣化碎屑的滲漏。指骨吸收 (phalangeal reabsorption) 與皮膚鈣質沉著症的合併,據說是系統性硬化症的病理特徵 (pathognomonic)。皮膚鈣質沉著症也曾在無硬皮病的系統性硬化症 (systemic sclerosis sine scleroderma) 病人中被報告。
肺部 X 光片典型上顯示雙側基底纖維化 (bilateral basal fibrosis),呈瀰漫性斑點狀或線性浸潤;囊腫形成(「蜂窩肺 honeycomb lung」)是不罕見的特徵。
心臟、腎臟、周邊神經、胃腸道與骨骼系統也會受侵犯:
- 大多數病人有亞臨床的原發性心臟侵犯。心臟侵犯可能表現為運動時呼吸困難、陣發性夜間呼吸困難 (paroxysmal nocturnal dyspnea)、心包炎 (pericarditis)、心包積液 (pericardial effusion)、充血性心衰竭、心律不整、瓣膜異常、心肌肥厚,或偶爾為非典型心絞痛。小冠狀動脈與小動脈的血管痙攣 (vasospasm) 已被觀察到為早期的心臟表現。包括心包炎與積液在內的顯著心臟異常是常見的病理發現,在屍檢中超過 50% 的個案中存在。然而,通常它們是無症狀的。偶爾,可能發展出嚴重的急性心包炎,且曾記錄到罕見的致命性心包填塞 (cardiac tamponade) 病例。大量心包積液與急性腎衰竭相關,並為不良的預後指標。斑塊狀的心肌瘢痕 (patchy myocardial scarring) 很常見,通常發生在系統性硬化症病程的較晚期。當嚴重時,心肌纖維化 (myocardial fibrosis) 與不良預後相關。它發生
系統性硬化症有時與一種類似結節性紅斑 (erythema nodosum-like) 的脂膜炎症候群 (panniculitis syndrome) 相關,病人也可能發展出影響下肢的網狀青斑 (livedo reticularis) 與白色萎縮 (atrophie blanche)。
CREST 曾被記錄到與家族性硬化性苔癬 (familial lichen sclerosus)、慢性骨髓性白血病 (chronic myelogenous leukemia)、特發性骨髓纖維化 (idiopathic myelofibrosis) 與遲發性皮膚紫質症 (porphyria cutanea tarda) 相關。
瀰漫型系統性硬化症 (Diffuse systemic sclerosis) 在瀰漫型(進行性)系統性硬化症中,皮膚病灶特別常見於近端肢體、胸部與腹部。病程傾向於進行性,且常有較嚴重的淺層血管侵犯。影響軀幹的皮膚增厚指示不良預後,並與廣泛的全身性侵犯相關。雖然皮膚特徵造成相當大的痛苦,但就嚴重的發病率與潛在死亡率而言,全身性表現更為重要。
獨立於冠狀動脈疾病之外,並曾在屍檢系列中高達 70% 的病人中被描述。主要冠狀動脈是通暢且正常的(除非同時存在動脈粥狀硬化),但小血管與小動脈可能發生內皮與內膜增生性變化伴瘢痕形成,導致心律不整風險增加與隨之而來的病人猝死。近期一項研究發現心肌灌注缺損 (myocardial perfusion defects)、皮膚厚度、手指潰瘍與食道侵犯之間有關聯。
-
腎臟侵犯表現為「硬皮病腎危象 (scleroderma renal crisis)」是一種極為重要的併發症,死亡率高,發生於約 10% 的系統性硬化症病人。然而,ACE inhibitors 的使用已顯著降低死亡率。它被定義為「新發生的加速性動脈高血壓和/或快速進行性少尿性腎衰竭」。病人發展出頭痛與視力模糊。癲癇發作有時是其特徵。腎衰竭常為無症狀,僅能從異常的腎功能檢查偵測到,包括蛋白尿、伴管型的顯微鏡下血尿、肌酸酐 (creatinine) 濃度升高,以及高腎素血症 (hyperreninemia)。微血管病性溶血性貧血 (microangiopathic hemolytic anemia) 有時存在,尤其在血壓正常的病人。Anti-RNA polymerase III 抗體已在約三分之一的「硬皮病腎危象」病人中被偵測到。少數系統性硬化症病人發展出「硬皮病腎危象」以外的腎臟病理。偶爾曾報告 ANCA 相關的腎絲球腎炎 (ANCA-related glomerulonephritis)。
-
周邊神經病變 (Peripheral neuropathy) 可能導致神經病變性潰瘍 (neuropathic ulceration)。曾描述患有此病的孕婦發生子宮內胎兒死亡 (intrauterine fetal death)。丘疹與結節性黏液病 (papular and nodular mucinosis) 曾被記錄為系統性硬化症的表現徵象。
-
臨床上相關的胃腸道病灶發生於高達 50% 的系統性硬化症病人。經放射學確定的牙周間隙增寬 (widening of the periodontal space) 是其特徵。病人常有與食道侵犯相關的症狀,包括胃灼熱、吞嚥困難 (dysphagia) 與逆流 (regurgitation)。胃腸逆流很常見,病人可能發展出食道炎 (esophagitis)、出血、狹窄、Barrett 食道(胃化生,gastric metaplasia)與吸入 (aspiration)。X 光片可能顯示食道擴張與蠕動異常。上腹部飽脹感是常見的症狀,可能與胃竇 (gastric antrum) 擴張受限有關。胃竇血管擴張 (gastric antral vascular ectasia) 似乎在那些呈現快速進行性皮膚疾病的系統性硬化症病人中較早發展。系統性硬化症常侵犯小腸,症狀範圍從上腹痛、噁心與嘔吐,到假性腸阻塞 (pseudo-obstruction) 的影響;由於滯留 (stasis) 所致的吸收不良狀態是重要的併發症。乳糜瀉 (celiac disease) 可在系統性硬化症病人中發展。結腸病灶可能導致腹瀉或便秘。沿結腸腸繫膜緣的囊狀憩室 (saccular diverticula) 是其特徵;它們有時也影響小腸。
-
骨關節侵犯,表現為關節痛或明顯的關節炎,見於大多數病人。關節病灶通常輕微,影響腕、手、膝與踝,雖然曾記錄到較嚴重的類風濕性關節炎樣 (rheumatoid arthritis-like) 變異型。骨關節病 (osteoarthrosis) 與類乾癬性關節病 (psoriatic arthropathy-like) 表現也曾被描述。對有顯著關節表現的病人,重要的是排除重疊症候群與 MCTD。導致活動不能的攣縮與關節僵硬 (ankyloses) 是重要的併發症,且由於不動 (immobilization) 與缺血的合併,骨質疏鬆很常見。系統性硬化症的診斷可能很容易明顯,但早期疾病(尤其是瀰漫型)在臨床上可能類似多種其他疾病,例如 Buschke 硬腫病 (scleredema of Buschke)。晚期移植物對抗宿主病 (graft-versus-host disease, GVHD) 與遲發性皮膚紫質症的慢性病灶典型上呈硬皮病樣 (sclerodermatous)。美國風濕病學會 (American Rheumatology Association) 有分類指引,其主要標準為近端硬皮病 (proximal scleroderma)(表 17.7)。次要標準為:
-
指趾硬化 (sclerodactyly),
-
指尖的指狀凹陷性瘢痕 (digital pitting scars) 或末節指腹物質喪失,
-
雙側基底肺纖維化 (bilateral basal pulmonary fibrosis)。
-
近端硬皮病為單一主要標準:敏感度為 91%,特異度超過 90%。
-
在無近端硬皮病的情況下,指趾硬化、指尖的指狀凹陷性瘢痕或指腹物質喪失,以及雙側基底肺纖維化構成進一步的次要標準。
-
主要標準或兩項以上次要標準存在於 97% 確定的系統性硬化症病人中,但僅存在於 2% 的對照病人(系統性紅斑性狼瘡、多發性肌炎–皮肌炎或雷諾病)。
*系統性硬化症的初步臨床標準排除局限性硬皮病與假硬皮病樣疾患。經許可重製自 Masi, A.T. et al. 與 the ARA Subcommittee for scleroderma (1980) Arthritis and Rheumatism, 23, 581–590。
若病人具有主要標準或兩項次要標準,則對確定的系統性硬化症有 97% 的敏感度與 98% 的特異度。這些標準已廣受歡迎,但有一個缺點,即排除了至少 10% 的個案——這些個案儘管有確切的系統性硬化症診斷,卻既不符合主要標準也不符合次要標準。無法分類的症候群與重疊症候群也被排除。其他分類根據皮膚硬化的範圍納入了兩種、三種,甚至四種亞型(表 17.8)。因此,局限性皮膚系統性硬化症可能侵犯手、足、前臂與顏面,或皮膚病灶可缺如,而在瀰漫型疾病中軀幹皮膚也會受侵犯。在以兩種亞型(瀰漫型與局限型)或三種亞型(瀰漫型、中間型與局限型)分類的比較中,後者與抗體特異性及存活的相關性最佳。
2013 年,美國風濕病學會 (American College of Rheumatism, ACR) 與歐洲抗風濕病聯盟 (European League Against Rheumatism, EULAR) 提出系統性硬化症分類標準的修訂,以提高敏感度,尤其在診斷早期型態的系統性硬化症與局限性皮膚系統性硬化症方面。新的 2013 ACR/EULAR 系統性硬化症分類標準採用一套計分系統,需要 ≥ 9 分才能將病人分類為患有系統性硬化症(表 17.9)。簡言之,根據新標準,手指皮膚增厚延伸至掌指關節近端被視為診斷系統性硬化症的充分標準(計 9 分)。或者,採用七項具不同權重的額外參數:
- 手指皮膚增厚(臃腫手指 2 分或指趾硬化 4 分,僅計較高分數),
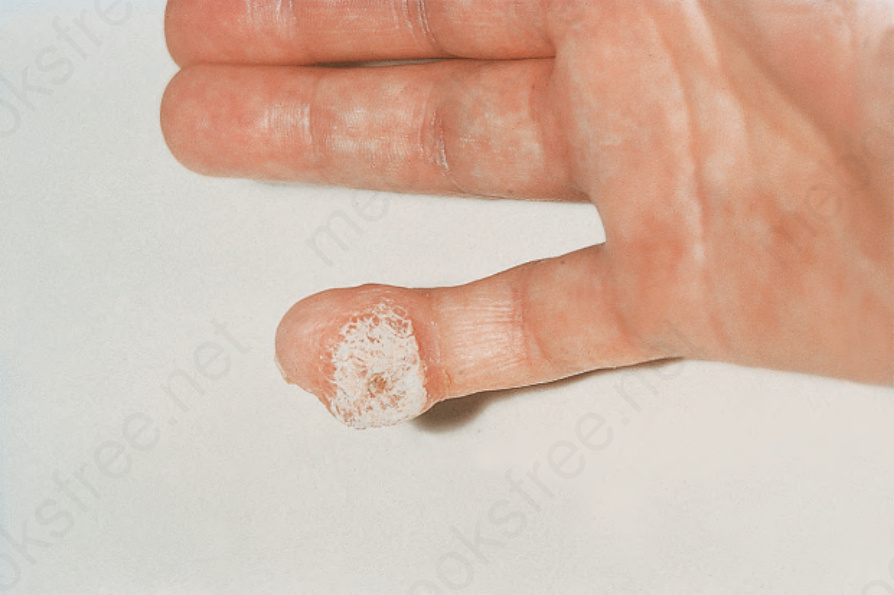
圖 17.93:系統性硬化症 (systemic sclerosis):指尖上有一個大的鈣化結節 (calcified nodule)。承蒙 R.A. Marsden, MD, St George’s Hospital, London, UK 提供。
Fig. 17.93 Systemic sclerosis: there is a large calcified nodule on the fingertip. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.

表 17.7:美國風濕病學會 (American Rheumatology Association, ARA) 硬皮病分類指引
Table 17.7 American Rheumatology Association (ARA) guidelines for the classification of scleroderma*
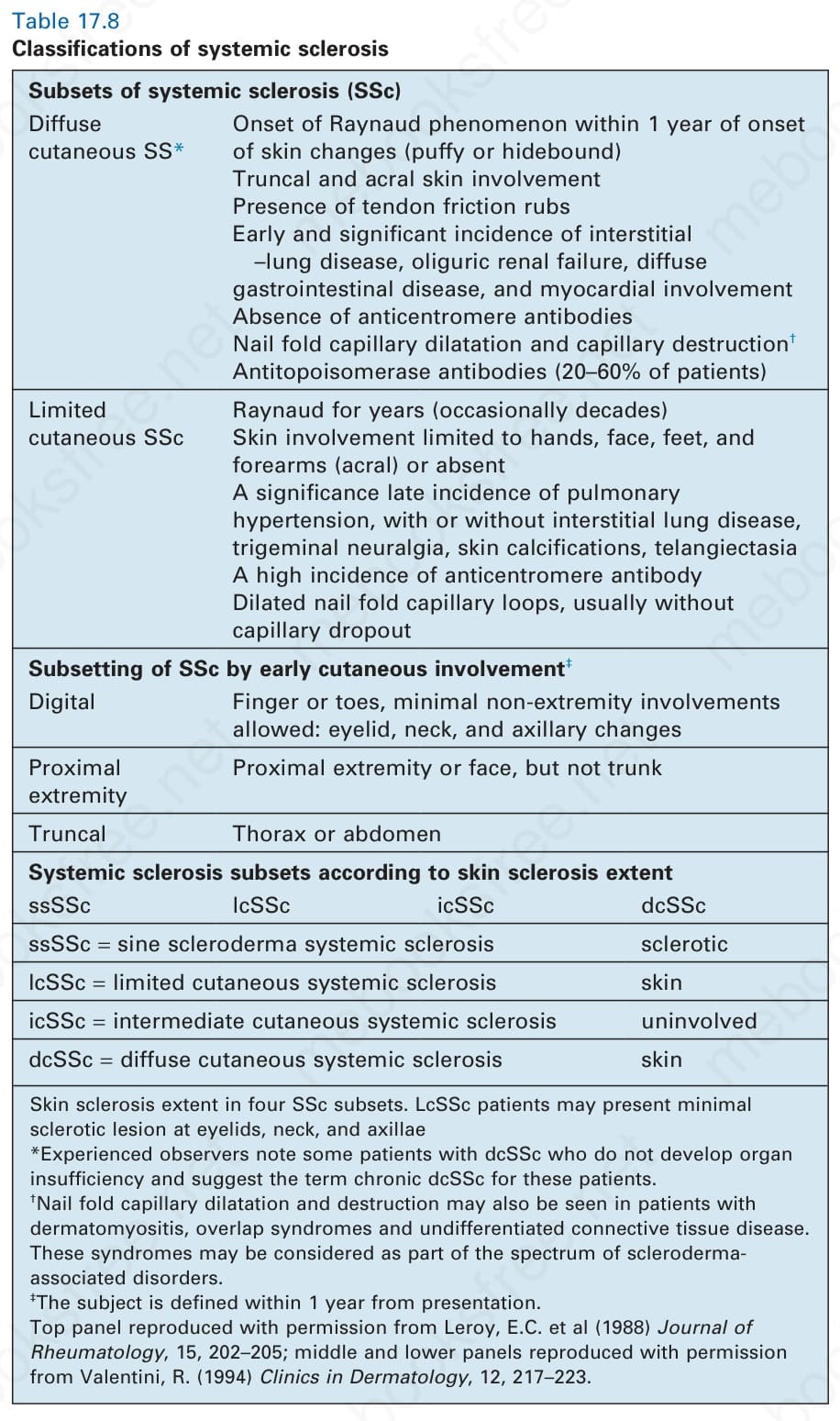
表 17.8:系統性硬化症的分類 (Classifications of systemic sclerosis)
Table 17.8 Classifications of systemic sclerosis
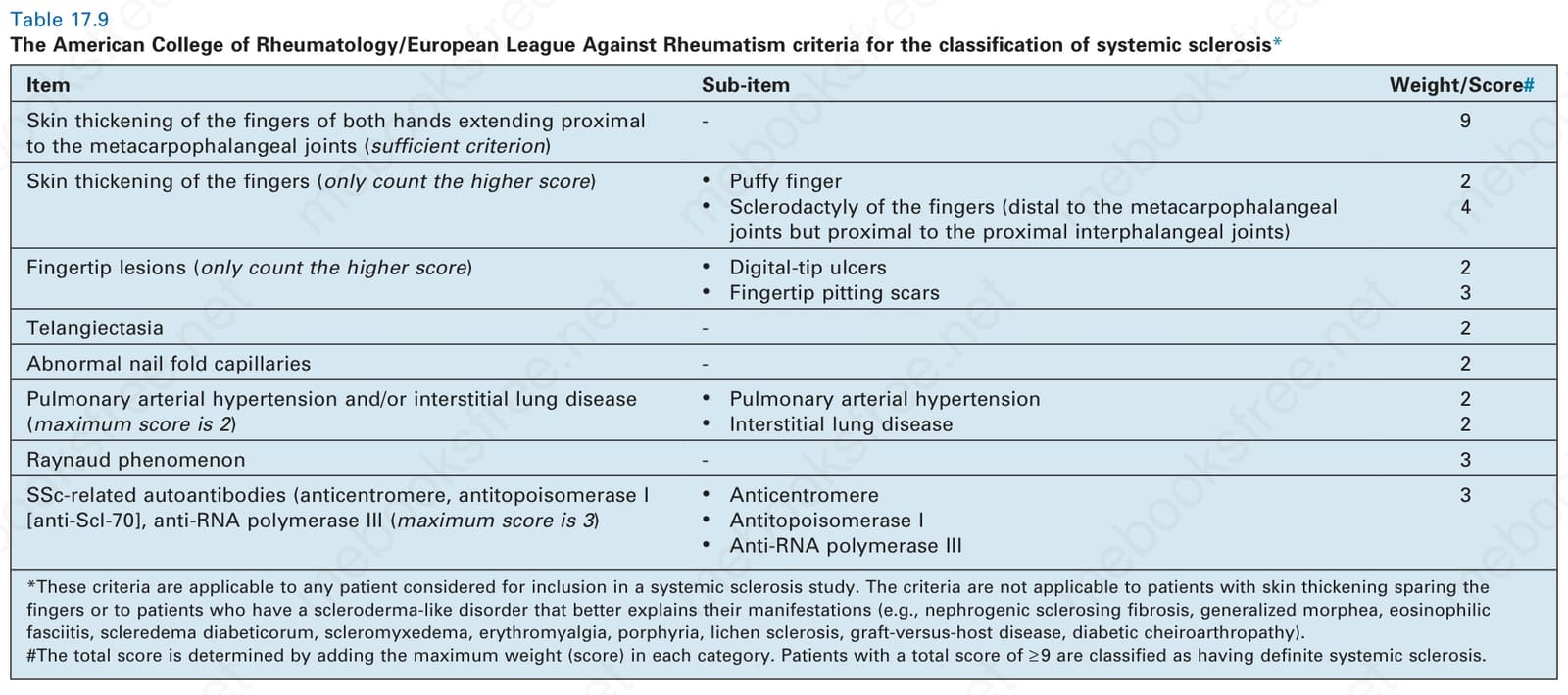
表 17.9:美國風濕病學會/歐洲抗風濕病聯盟 (American College of Rheumatology/European League Against Rheumatism) 系統性硬化症分類標準
Table 17.9 The American College of Rheumatology/European League Against Rheumatism criteria for the classification of systemic sclerosis*
-
指尖病灶(指尖潰瘍 2 分或凹陷性瘢痕 3 分,僅計較高分數),
-
毛細血管擴張(2 分),
-
異常甲褶微血管(2 分),
-
間質性肺病或肺動脈高壓 (pulmonary arterial hypertension)(2 分),
-
雷諾現象(3 分),以及
-
系統性硬化症特異性自體抗體(3 分)。
藉由應用這套新標準,一項來自加拿大、納入 724 名系統性硬化症病人的大型研究發現,新的 2013 標準的整體敏感度為 98.3%,相較於 1980 標準的 88.3%,顯著改善了早期系統性硬化症與局限性皮膚系統性硬化症的診斷準確度。此外,近期一項納入 3196 名系統性硬化症病人的研究顯示,手指潰瘍病史不僅對新發手指潰瘍的發展具有預測價值,也對肺動脈壓升高與其他可能的心血管事件具有預測價值,並且代表存活率降低的一個獨立預測因子。
已描述若干化學物質誘發的硬皮病樣症候群:
- 氯乙烯 (vinyl chloride) 聚合工業的工作者可能發展出雷諾現象、肢端骨溶解 (acral osteolysis)、手臂、雙手、顏面與軀幹皮膚的真皮增厚,以及肺與肝纖維化。甲褶微血管的檢查顯示與系統性硬化症所見類似的異常。
系統性硬化症 (SSc) 的亞群 瀰漫型皮膚 SSc*
雷諾現象於皮膚變化(臃腫或緊束 hidebound)發作後 1 年內發作;軀幹與肢端皮膚侵犯;肌腱摩擦音的存在;間質性肺病、少尿性腎衰竭、瀰漫性胃腸道疾病與心肌侵犯的早期且顯著發生率;無抗著絲點抗體;甲褶微血管擴張與微血管破壞†
舌鱗狀細胞癌 (squamous cell carcinoma of the tongue) 的發生率增加也已被偵測到。罕見的相關包括基底細胞癌 (basal cell carcinoma)、黑色素瘤 (melanoma)、鼻咽癌 (nasopharyngeal carcinoma)、胃 MALT 淋巴瘤 (gastric MALT lymphoma)、膀胱癌、子宮頸癌、食道癌、甲狀腺癌、皮膚鱗狀細胞癌,以及骨髓增生不良症候群 (myelodysplastic syndrome)。
致病機轉與組織學特徵 (Pathogenesis and histologic features) 病因與精確的致病機轉不明。完整的理解必須考量血管變化與膠原蛋白沉積及分布的異常,此外還有那些在早期階段具特徵性的發炎細胞之意義,以及它們在纖維母細胞 (fibroblast) 生長與功能調控中的角色。系統性硬化症已激發了龐大的研究努力,這使人們更加意識到可能涉及的多種因子(無論是單獨或協同作用),並大幅增加了我們對膠原蛋白合成與瘢痕形成機制中所涉基本過程的知識。兩個主要的研究領域圍繞著:
- 原發性血管內皮細胞損傷及其後遺症,
- 膠原蛋白及其合成的異常。
抗拓樸異構酶抗體 (Antitopoisomerase antibodies)(20–60% 的病人)
局限型皮膚 SSc (Limited cutaneous SSc)
雷諾現象持續多年(偶爾數十年);皮膚侵犯局限於雙手、顏面、足部與前臂(肢端 acral)或缺如;肺高壓的顯著晚期發生率(無論是否伴間質性肺病)、三叉神經痛 (trigeminal neuralgia)、皮膚鈣化、毛細血管擴張;抗著絲點抗體的高發生率;擴張的甲褶微血管環,通常無微血管脫落 (capillary dropout)
依早期皮膚侵犯對 SSc 進行亞分類‡
指趾型 (Digital):手指或腳趾,容許極少的非肢端侵犯:眼瞼、頸部與腋窩變化
兩者皆固有的,是細胞媒介性 (cell-mediated) 與體液性免疫 (humoral immunity) 可能的起始與調節角色。
長久以來已認識到系統性硬化症的許多特徵可能有缺血的基礎。已描述微血管、小靜脈與動脈的改變,且有人認為初始損傷涉及微血管內皮細胞。其原因不明,雖然已鑑定出一種對內皮細胞具反應性的循環特異性細胞毒性物質。有人認為這可能代表一種蛋白酶 (protease)。有趣的是,早期病灶與未受侵犯皮膚的檢體已顯示內皮細胞損傷的超微結構證據,合併氚標記腺苷 (tritiated adenosine) 攝取減少與可免疫偵測的 von Willebrand factor 儲存減少,顯示血管變化很可能起始此病所見的結締組織損傷。
近端肢體 (Proximal extremity)
近端肢體或顏面,但非軀幹
軀幹型 (Truncal):胸部或腹部
依皮膚硬化範圍區分的系統性硬化症亞群 ssSSc lcSSc icSSc dcSSc
ssSSc = 無硬皮病系統性硬化症 (sine scleroderma systemic sclerosis) 硬化的
lcSSc = 局限型皮膚系統性硬化症 (limited cutaneous systemic sclerosis) 皮膚
icSSc = 中間型皮膚系統性硬化症 (intermediate cutaneous systemic sclerosis) 未受侵犯
dcSSc = 瀰漫型皮膚系統性硬化症 (diffuse cutaneous systemic sclerosis) 皮膚
四種 SSc 亞群的皮膚硬化範圍。LcSSc 病人可能在眼瞼、頸部與腋窩呈現極少的硬化病灶。*有經驗的觀察者注意到一些 dcSSc 病人並未發展出器官功能不全,並建議對這些病人使用慢性 dcSSc (chronic dcSSc) 一詞。†甲褶微血管擴張與破壞也可見於皮肌炎、重疊症候群與未分化結締組織病的病人。這些症候群可被視為硬皮病相關疾患譜系的一部分。‡此主題於發病後 1 年內定義。上面板經許可重製自 Leroy, E.C. et al (1988) Journal of Rheumatology, 15, 202–205;中與下面板經許可重製自 Valentini, R. (1994) Clinics in Dermatology, 12, 217–223。
雖然透過免疫螢光技術已在腎絲球微血管壁中偵測到免疫反應物 (immunoreactants)(IgG 與補體),但在皮膚血管中並未發現它們。若於系統性硬化症病人誘發雷諾現象,會伴隨腎臟與肺臟血流的同時減少,暗示有一個尚未鑑定的循環因子。
-
Bleomycin 治療也可能與硬皮病樣 (sclerodermiform) 的浸潤性斑塊與結節相關,這些病灶特別影響雙手。病人可能發展出色素沉著過度、周邊壞疽與肺纖維化。
-
在煤礦工作或因其他原因過度暴露於矽 (silica) 者中,發現系統性硬化症的高發生率。
-
一種全身性硬斑病樣 (morphea-like) 變異型,伴隨雷諾現象、食道功能障礙與肺纖維化,曾被描述發生於慢性暴露於工業溶劑之後。
-
在使用矽膠隆乳 (silicone breast implants) 之後曾記錄到多種自體免疫疾病。系統性硬化症似乎是最常見的。
-
毒油症 (Toxic oil) 與嗜伊紅性肌痛症候群 (eosinophilia-myalgia syndromes) 將在嗜伊紅性筋膜炎 (eosinophilic fasciitis) 的章節中討論。系統性硬化症與惡性腫瘤風險增加相關,惡性腫瘤發展於 3.6% 到 10.7% 的病人。以族群為基礎的研究發現,最常見的相關為乳癌、肺癌與血液惡性腫瘤,如非何杰金氏淋巴瘤 (non-Hodgkin lymphoma)。增加的
真皮微血管顯示多種超微結構變化。最早的發現是內皮細胞的分離;這可能導致液體滲漏,因此至少部分地造成早期階段特有的水腫。更嚴重損傷的證據表現為內皮細胞空泡化 (vacuolation)、中間絲 (intermediate filaments) 數目增加、胞飲囊泡 (pinocytotic vesicles) 與 Weibel-Palade 小體減少,以及異常的內皮表面細胞質泡 (cytoplasmic blebs) 的存在。
內皮細胞損傷的證據可藉由估計血漿 von Willebrand factor 濃度於臨床上監測。系統性硬化症典型上可見超正常的 von Willebrand factor 多聚體 (multimers) 濃度升高,並可能具有致病意義,因為已知它們會結合至內皮下組織,造成血小板聚集與黏附,導致血管增生與血栓形成。ACE 濃度已證實在系統性硬化症中降低,這在評估內皮細胞損傷的存在上也可能有價值。內皮細胞衍生的胜肽——內皮素 (endothelin)(會造成血管收縮)的濃度升高已被鑑定。內皮素也具有纖維母細胞促分裂活性 (mitogenic activity) 並刺激膠原蛋白的合成。
終末期表現為微血管壁的完全破壞;細胞核呈顆粒狀且均質,細胞膜破裂,細胞質內容物見於微血管管腔與血管外間隙。內皮細胞對氚標記腺苷的攝取已證實減少。基底膜增厚與重複 (reduplication) 常存在,血管周圍纖維化是常見的晚期伴隨現象。血管損傷的最終結果最容易藉由甲褶微血管鏡來證明。殘餘血管的擴張很可能代表一種代償措施。這些殘餘血管中內皮細胞增生的增加已藉由氚標記胸腺嘧啶核苷 (tritiated thymidine) 攝取研究獲得證實。
項目 子項目 權重/分數#
雙手手指皮膚增厚延伸至掌指關節近端(充分標準)
- 9
手指皮膚增厚(僅計較高分數)
- 臃腫手指 (Puffy finger)
- 手指指趾硬化(掌指關節遠端但近端指間關節近端)
2 4
指尖病灶(僅計較高分數)
- 指尖潰瘍 (Digital-tip ulcers)
- 指尖凹陷性瘢痕 (Fingertip pitting scars) 2 3
毛細血管擴張 (Telangiectasia) - 2
異常甲褶微血管 - 2
- 肺動脈高壓 (Pulmonary arterial hypertension)
- 間質性肺病 (Interstitial lung disease) 2 2
肺動脈高壓和/或間質性肺病(最高分為 2)
雷諾現象 (Raynaud phenomenon) - 3
- 抗著絲點 (Anticentromere)
- 抗拓樸異構酶 I (Antitopoisomerase I)
- Anti-RNA polymerase III
SSc 相關自體抗體(anticentromere、antitopoisomerase I [anti-Scl-70]、anti-RNA polymerase III(最高分為 3)
3
*這些標準適用於任何考慮納入系統性硬化症研究的病人。這些標準不適用於皮膚增厚不侵犯手指的病人,或具有能更好解釋其表現之硬皮病樣疾患(例如腎源性硬化性纖維化 nephrogenic sclerosing fibrosis、全身性硬斑病 generalized morphea、嗜伊紅性筋膜炎 eosinophilic fasciitis、糖尿病性硬腫病 scleredema diabeticorum、硬化性黏液水腫 scleromyxedema、紅斑性肢痛症 erythromyalgia、紫質症 porphyria、硬化性苔癬 lichen sclerosis、移植物對抗宿主病 graft-versus-host disease、糖尿病性手關節病 diabetic cheiroarthropathy)的病人。#總分藉由加總每一類別中的最高權重(分數)來決定。總分 ≥ 9 的病人被分類為患有確定的系統性硬化症。
小動脈也涉及血管破壞現象,特徵為血管壁增厚,由平滑肌增生、纖維化與過量糖胺聚醣 (glycosaminoglycans) 沉積的合併所致。動脈顯示非常顯著的內膜增厚,這在腎臟弓狀血管 (arcuate vessels) 中特別清楚可見,常因同心性層狀結構而被稱為「洋蔥皮樣 (onion skinning)」。它是由於黏液樣變化 (myxoid change)、細胞增生與纖維化的結果而發展。
系統性硬化症病人皮膚中的大多數發炎細胞為 CD4+ T 細胞。
許多細胞激素 (cytokines) 已與系統性硬化症的致病機轉相連結。轉化生長因子 β (transforming growth factor beta, TGF-β) 與 IL-4 增加纖維母細胞增生與膠原蛋白合成,可能在此病的纖維化誘導中很重要。IL-17 是一種由 T 細胞分泌的細胞激素,可活化並誘導纖維母細胞增生與內皮細胞活化。此細胞激素已證實在受影響病人的皮膚與血液中增加,尤其在疾病的早期階段。它活化纖維母細胞分泌促發炎細胞激素 IL-6 與 -8,並增加細胞間黏附分子-1 (intercellular adhesion molecule-1, ICAM-1) 的表面表現。IL-17 也活化內皮細胞分泌 IL-6 與 -1,並表現 ICAM-1 與血管細胞黏附分子-1 (vascular cell adhesion molecule-1, VCAM-1)。IL-6 也能誘導纖維母細胞增生與膠原蛋白合成。IL-17 與其所誘導的其他細胞激素之合併效應,導致微循環的損傷以及皮膚與內臟的纖維化。有人認為結締組織生長因子 (connective tissue growth factor)(其產生由 TGF-β 誘導)可能在纖維化的致病機轉中扮演重要角色。近期已證實 CD8+ 效應 T 淋巴球是系統性硬化症病人血清中 IL-13 增加的來源。IL-13 具有眾所周知的促纖維化活性,藉由直接刺激纖維母細胞,並間接藉由刺激 TGF-β。TGF-β 除了纖維生成 (fibrogenesis) 外,也藉由誘導內皮素的合成(內皮素作為一種強效的血管收縮劑,並已被牽涉於潰瘍的致病機轉)而促成系統性硬化症血管異常的發展。
特異性,它在不同族群之間有所變化。美洲原住民與日本病人有高頻率的 anti-fibrillin-1 抗體。
在患有系統性硬化症的女性的多個器官中發現了男性細胞,但在健康女性中則無。胎兒細胞遷移至母體循環並在不同器官中存活,被稱為微嵌合現象 (microchimerism)。微嵌合現象在系統性硬化症的致病機轉中扮演什麼角色(如果有的話)仍不清楚。
系統性硬化症主要的組織學特徵是瘢痕形成。密集的研究已確認膠原蛋白量增加的存在,但精確的致病機轉至今仍不確定。在系統性硬化症病人中已證實脯胺酸羥化酶 (proline hydroxylase) 活性增加與標記脯胺酸 (proline) 攝取增加,這兩者皆為活躍膠原蛋白合成的指標。典型上可見可還原醛亞胺交聯 (reducible aldimine cross-links) 的濃度升高,這是新合成膠原蛋白的特徵。第三型膠原蛋白 N 端前胜肽 (N-terminal propeptide of type III collagen) 的血清濃度升高與羥脯胺酸 (hydroxyproline) 尿排泄增加也已被記錄。
來自系統性硬化症病人的纖維母細胞培養所合成的膠原蛋白比來自正常對照組者更多。雖然曾報告組織膠原蛋白酶 (collagenase) 濃度降低,但其他研究者並未證實此發現,因此其意義不確定。膠原纖維的胺基酸組成正常。演變中病灶的電子顯微鏡檢查已揭示未成熟膠原原纖維 (collagen fibrils) 的存在,特徵為狹窄的口徑(30 nm)、未成熟的條帶型態,以及雙股珠狀絲 (double-stranded beaded filaments)。在較成熟的病灶中,膠原纖維接近正常厚度(100 nm),但其分布高度無序。Luse 小體有時是其特徵。
系統性硬化症中的纖維母細胞能夠組裝微原纖維 (microfibrils),但這些不穩定(可能由於 fibrillin 1 此一細胞外基質蛋白的內在缺陷),這也可能在此病的致病機轉中扮演角色。有趣的是,fibrillin-1 基因的重複已被牽涉為緊皮 (tight skin) 的原因,而緊皮是系統性硬化症的一種動物模型。在系統性硬化症病人的血清中可見抗 fibrillin 抗體升高。雖然這似乎具高度疾病特異性,它在不同族群之間有所變化。美洲原住民與日本病人有高頻率的 anti-fibrillin-1 抗體。
患有瀰漫型與局限型皮膚系統性硬化症的病人,與健康對照組相比,可溶性 CD163 (soluble CD163) 的平均血清濃度升高。近期已證實,血清可溶性 CD163 濃度升高的系統性硬化症病人,其肺動脈收縮壓比血清 CD163 濃度正常者更高,暗示巨噬細胞可能在系統性硬化症的致病機轉中扮演角色。相反地,藉由結合至一種 TNF 樣弱凋亡誘導物 (TNF-like weak inducer of apoptosis),CD163 可能保護免於手指潰瘍的發展,但卻促成更顯著的皮膚纖維化。
可溶性 T 細胞免疫球蛋白與黏蛋白結構域 3 (soluble T-cell immunoglobulin and mucin domain 3, TIM-3) 的血清濃度,在瀰漫型皮膚系統性硬化症病人中比在局限型皮膚系統性硬化症病人與健康個體中更高,且與皮膚硬化的嚴重度呈正相關,尤其在瀰漫型皮膚系統性硬化症的
早期階段。TIM-3 的血清濃度升高也與心臟侵犯及腎危象相關。
活躍病灶的組織學檢查常顯示纖維母細胞數目增加。已顯示來自下層真皮的纖維母細胞所合成的膠原蛋白比來自上層真皮者更多,暗示系統性硬化症中有兩種不同的細胞群。纖維化(由於第 I、III、V 與 VI 型膠原蛋白的沉積所致)伴隨過量的纖維連接蛋白 (fibronectin)。
關於 anti-fibrillarin (anti-U3-RNP) 自體抗體的研究確認了其與較年輕的發病年齡、男性、非裔加勒比血統 (Afro-Caribbean descent)、較高的 Rodnan 皮膚評分嚴重度指數,以及肌炎相關,但與瀰漫型皮膚系統性硬化症的存在、肺侵犯或存活差異無關。Anti-U11/U12-RNP 抗體已在約 3% 的系統性硬化症中被證實,並具有肺纖維化與胃腸道侵犯的風險增加。Anti-Ku 抗體在 2.2% 的系統性硬化症中可偵測到,並與肌肉骨骼異常(如肌炎、關節炎與關節攣縮)以及指尖潰瘍與毛細血管擴張相關。
近期,在受侵犯皮膚的真皮內也已證實大量的第 VII 型膠原蛋白,伴隨 TGF-β 表現升高。已知後者會上調第 VII 型膠原蛋白基因的活性。此發現具有潛在的重要性,因為第 VII 型膠原蛋白的分布通常局限於真皮–表皮交界處的錨定原纖維 (anchoring fibrils)。第 I 與第 III 型膠原蛋白 mRNA 表現增加已在來自硬皮病病人的培養纖維母細胞中被證實。系統性硬化症的特徵為每單位重量的膠原蛋白濃度正常。然而相反地,每單位表面積的膠原蛋白含量大幅增加。
膠原蛋白合成具有負回饋控制。因此,在前膠原蛋白 (procollagen) 分子的胺基端被胺基端胜肽酶 (amino terminal peptidase) 切割後,釋放出的胺基端會抑制膠原蛋白的形成。藉由免疫電子顯微鏡技術已顯示,在膠原原纖維的部位有胺基胜肽 (amino peptide) 的滯留。
有若干抗核抗體 (antinuclear antibodies) 的亞群,它們也具有臨床預測價值:
-
抗著絲點抗體(幾乎對系統性硬化症具特異性)在局限型皮膚變異型中特別常見。它通常見於疾病較不嚴重且結果較有利的病人。鈣質沉著與毛細血管擴張可能很明顯,但間質性肺纖維化的可能性較低。
-
Scl-70 抗體 (anti-DNA topoisomerase) 見於 20% 到 60% 的系統性硬化症病人,尤其是瀰漫型變異型。它也具高度特異性。Scl-70 抗體與嚴重的全身性侵犯(包括肺間質纖維化)及不良預後相關。
-
抗中心粒抗體 (Anticentriole antibody) 出現於局限型與瀰漫型兩種型態。雖然這些抗體具有重大的診斷重要性,但它們似乎沒有任何致病意義。
除了膠原蛋白量增加外,系統性硬化症早期病灶的皮膚含有過量的糖胺聚醣,特別是硫酸皮膚素 (dermatan sulfate) 與硫酸軟骨素 4- 與 6-硫酸鹽 (chondroitin 4- and 6-sulfate)。有些證據顯示糖胺聚醣的增加可能至少部分地由於降解減少;它們的存在與活體內的水結合相關,因此推測也是造成水腫的原因。
系統性硬化症與體液性及細胞性免疫的異常相關。與 SLE 相反,anti-DNA 抗體通常不存在。然而,幾乎所有病人都具有抗核抗體;這些可能是斑點型 (speckled)、均質型 (homogeneous) 或核仁型 (nucleolar)(圖 17.95)。後者見於 7% 到 46% 的病人,但不具特異性,可見於若干其他結締組織病。它們形成一個異質性群組,對抗多種抗原,包括 U3-RNP (fibrillarin)、RNA polymerase I、Th ribonucleoprotein 與 PM-Scl,並具有一些預後意義與亞型特異性(表 17.10)。Anti-U3 RNP 抗體存在於 5% 到 8% 的系統性硬化症,並與非裔美國人種族、男性、較高的骨骼肌病理發生率,以及肺動脈高壓相關。一項進一步的研究
近期一項來自荷蘭、分析共 460 名病人的研究顯示,間質性肺病在具抗拓樸異構酶 I 抗體的局限性皮膚系統性硬化症病人中,比沒有這些抗體的病人更為盛行(分別為 49% 與 25%)。此外,具抗拓樸異構酶 I 抗體的局限性皮膚系統性硬化症病人更可能進展為瀰漫型皮膚系統性硬化症。
Ro (SS-A) 與 La (SS-B) 抗體的存在暗示 Sjögren 症候群的並存。對抗基質金屬蛋白酶-3 (matrix metalloproteinase-3) 的自體抗體已在約 50% 的系統性硬化症病人中被偵測到。它們在瀰漫型皮膚系統性硬化症病人中比在局限型皮膚系統性硬化症病人中顯著較高,並與皮膚纖維化、肺纖維化與腎血管增厚顯著相關。
對抗第 I 與第 IV 型膠原蛋白的抗體已被描述,但尚不清楚它們是代表原發性致病因子或繼發現象。較近期,抗內皮細胞抗體 (antiendothelial cell antibodies) 已在硬皮病中被記錄。循環免疫複合體 (circulating immune complexes) 已
圖 17.95:系統性硬化症 (systemic sclerosis):抗核仁抗體 (antinucleolar antibody)(HEP II)。承蒙 G. Swana, MD, St Thomas’ Hospital, London, UK 提供。
Fig. 17.95 Systemic sclerosis: antinucleolar antibody (HEP II). By courtesy of G. Swana, MD, St Thomas’ Hospital, London, UK.
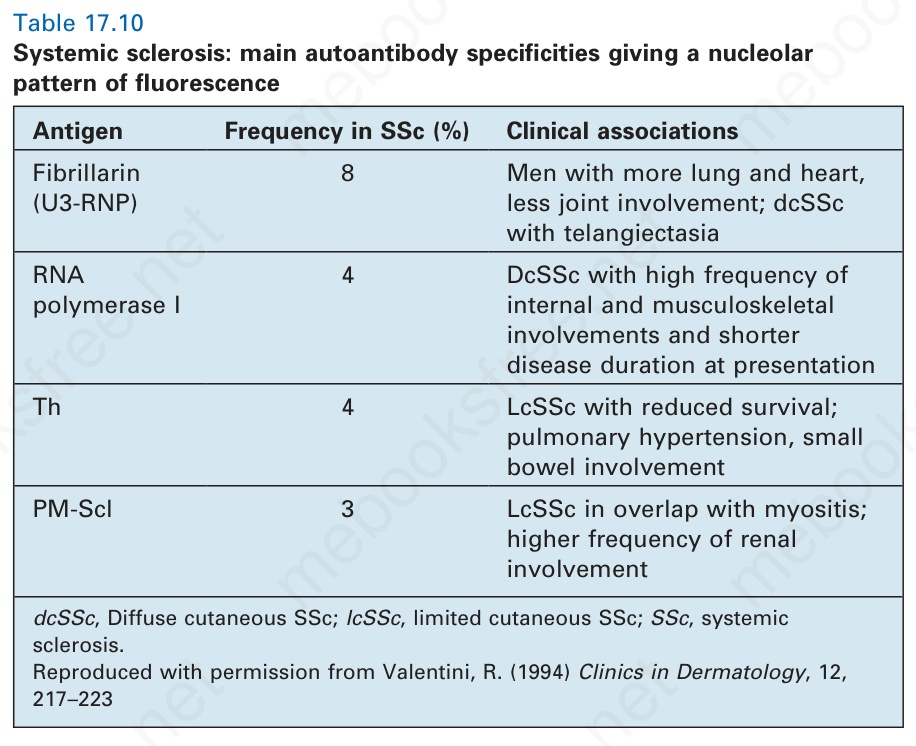
表 17.10:系統性硬化症 (systemic sclerosis):產生核仁型螢光型態的主要自體抗體特異性
Table 17.10 Systemic sclerosis: main autoantibody specificities giving a nucleolar pattern of fluorescence
抗原 在 SSc 中的頻率 (%) 臨床相關
Fibrillarin (U3-RNP)
8 男性,較多肺與心臟、較少關節侵犯;dcSSc 伴毛細血管擴張
RNA polymerase I
4 DcSSc,內臟與肌肉骨骼侵犯的高頻率,且發病時疾病病程較短
Th 4 LcSSc,存活降低;肺高壓、小腸侵犯
PM-Scl 3 LcSSc 與肌炎重疊;腎臟侵犯的頻率較高
dcSSc,瀰漫型皮膚 SSc;lcSSc,局限型皮膚 SSc;SSc,系統性硬化症。經許可重製自 Valentini, R. (1994) Clinics in Dermatology, 12, 217–223
已被報告,但並非恆定的特徵,其意義(如果有的話)不明。
已報告各種 T 細胞異常,其中大多數指向循環 T 淋巴球濃度減少,尤其是抑制亞群 (suppressor subset)。已描述輔助:抑制 (T helper:suppressor) 比率增加。可溶性細胞毒性 T 淋巴球相關分子-4 (soluble cytotoxic T lymphocyte-associated molecule-4, sCTLA-4) 已發現在瀰漫型皮膚系統性硬化症病人中增加,且 sCTLA-4 濃度升高似乎與疾病嚴重度與活性相關。T 細胞的克隆性擴增 (clonal expansion) 已在系統性硬化症病人的血液(61%)與皮膚(45%)中被偵測到,顯著高於正常配對對照組。
近期研究已朝向細胞激素在系統性硬化症纖維化發展中的角色。證據顯示它們是纖維母細胞功能與膠原蛋白合成的主要調節者。有人提出,在系統性硬化症中存在過度的纖維母細胞刺激活性,例如由於纖維母細胞趨化因子(包括纖維連接蛋白、膠原蛋白片段、血小板衍生生長因子 (platelet-derived growth factor)、表皮生長因子 (epidermal growth factor) 與 C5a)所致。纖維母細胞生長刺激因子(包括 IL-1、-2 與 -3、TGF-β 與血小板衍生生長因子)也具有重大重要性。
至今在系統性硬化症中尚未發現一致的強烈第 I 類或第 II 類主要組織相容性複合體 (major histocompatibility complex, MHC) 抗原相關。然而,個別自體抗體與 HLA 有顯著相關。因此,PM-Scl 抗體與 HLA-DR3 相關,而 Scl-70 抗體與 HLA-DR5 相關。
近期描述與系統性硬化症發展相關的易感基因包括 STAT-4、IRF5 與 BANK-1。
Fleischmajer 與 Lebwohl 提出了一個對系統性硬化症病理生理學有用的工作假說。他們提出,在血管損傷(可能由自體免疫機制所致)之後,第 IV 型膠原蛋白或其他物質的暴露導致 B 與 T 淋巴球以及單核球與肥大細胞的募集。過量的 T 輔助細胞刺激 B 細胞產生自體抗體,而活化的 T 細胞、巨噬細胞與肥大細胞分泌多種細胞激素,這些細胞激素轉而促進膠原蛋白沉積 (collagenosis)。
組織病理特徵 (Histopathology)
組織學上,早期階段的水腫產生一個與 Buschke 硬腫病無法區分的圖像。在已確立的病灶中,表皮有時顯得正常,或可能有表皮突 (rete ridge) 型態的喪失。基底細胞常有色素增加,且噬黑色素細胞 (melanophages) 在淺層真皮中常見。特徵性的變化是真皮被寬大、伸長、腫脹的膠原束所增厚,這些膠原束常呈現與表面上皮平行的方向排列(圖 17.96 與 17.97)。個別纖維的邊界常不清楚,使膠原蛋白呈現相當均質的外觀。彈性纖維通常不受影響。纖維化特徵性地侵犯皮下組織,因此脂肪細胞通常被併入真皮中。萎縮的皮膚附屬器(尤其是外分泌汗腺)是常見的特徵。
動脈(尤其是指趾血管)典型上顯示內皮細胞腫脹、內膜增厚與中膜肥厚 (medial hypertrophy)。之後它們可能變得透明化 (hyalinized)(圖 17.98)。在早期病灶中,內皮相關血小板的數目顯著增加。纖維蛋白 (fibrin) 沉積有時存在,偶爾完全阻塞導致指趾潰瘍與壞疽。隨著慢性化,血管數目進行性減少,尤其在較淺層的真皮。神經周圍纖維化 (perineural fibrosis) 有時是其特徵,鈣化並不罕見(圖 17.99 與 17.100)。
在早期病灶中,可能有慢性發炎細胞浸潤,由淋巴球、組織球與少數漿細胞 (plasma cells) 組成,位於血管周圍與真皮和皮下脂肪之間的交界處。T 輔助細胞佔優勢,且已描述真皮 Langerhans 細胞數目增加。肥大細胞(通常已活化)常以增加的數目存在。柵欄狀中性球與肉芽腫性皮膚炎 (palisaded neutrophilic and granulomatous dermatitis) 曾在一位局限型系統性硬化症病人中被報告。在一位 CREST 症候群病人的真皮與皮下組織中沉積澱粉樣蛋白 (amyloid) 也曾被報告。
通常無法在組織學基礎上區分局限性硬皮病(硬斑病,morphea)與系統性硬化症,雖然局限型的表皮通常正常且血管變化較不嚴重。相反地,發炎細胞浸潤在局限型變異型中常較濃重,且常影響網狀真皮。
骨骼肌的檢查可能顯示局灶性瘢痕與慢性發炎細胞浸潤(圖 17.101 與 17.102)。肌肉變性(空泡化、伴嗜伊紅性的均質化,以及橫紋喪失)與再生(嗜鹼性與肌膜核增生)的特徵——類似皮肌炎所見者——也可能存在。在硬化期則有萎縮與纖維化。
腎臟的巨觀檢查常顯示多發性梗塞、出血灶,偶爾有腎皮質壞死 (renal cortical necrosis) 的特徵。組織學外觀類似惡性高血壓 (malignant hypertension)
並以纖維素樣壞死 (fibrinoid necrosis) 的存在為特徵——其特別影響小葉間動脈與弓狀動脈——以及缺血性腎絲球硬化 (ischemic glomerulosclerosis);發炎細胞浸潤並非其特徵。系統性硬化症的特徵是小葉間動脈內膜中水腫與黏液樣變化 (mucoid change) 的存在。也有細胞增多 (increased cellularity),產生特徵性的「洋蔥皮 (onion skin)」外觀,伴血管管腔的縮小。
硬皮病性間質性肺炎的組織學特徵與特發性纖維化肺泡炎 (idiopathic fibrosing alveolitis) 所見者無法區分。早期階段的特徵為肺泡內水腫,伴反應性肺細胞 (reactive pneumocytes) 與不定量的巨噬細胞、淋巴球及偶爾中性球的浸潤。可見淋巴球與漿細胞的間質聚集,有時與局灶性淋巴樣增生 (focal lymphoid hyperplasia) 相關;在較舊的病灶中,這些伴隨富含糖胺聚醣的新膠原蛋白的沉積。終末期疾病的特徵為發展出大小不一的囊腫,由化生性細支氣管上皮 (metaplastic bronchiolar epithelium) 襯覆,其壁內含大量膠原蛋白與增生的平滑肌。
已發現輕度的內臟侵犯,包括食道下括約肌壓力異常與蠕動衰竭,以及高達 19% 病人的一氧化碳擴散輕微受損。這些異常並未導致臨床症狀,也不會不利地影響預後。曾記錄一例罕見病例,其中硬斑病誘發嚴重的肺外胸廓限制。皮膚外侵犯存在於約五分之一的兒童,並包括關節、神經、血管、眼、胃腸道、呼吸、心臟與腎臟表現,依頻率遞減順序排列。雖然斑塊以及程度較輕的線狀病灶常隨時間改善,但攣縮與半側萎縮 (hemiatrophy) 是永久性的。影像研究常顯示肌肉萎縮與腿長差異。局限性硬皮病可能發生於外傷、腹腔鏡檢查 (laparoscopy)、放射治療、刺青與矽膠植入之後。它也曾被描述與 bromocriptine、balicatib、valproic acid 與 ibuprofen 治療相關。局限性硬皮病也曾被報告發生於矽塵 (silica dust) 暴露之後。
肺高壓的特徵常存在,尤其在 CREST 變異型的病人。主要影響肌性小動脈 (muscular arterioles),雖然在晚期階段小靜脈也可能受侵犯,並顯示中膜肌肉肥厚與內膜中富含黏液樣的新膠原蛋白同心性沉積,伴管腔直徑的不定量縮小。在晚期階段,可能明顯出現肌肉萎縮與中膜彈性纖維變性 (medial elastosis)。曾記錄局灶性淋巴球/漿細胞血管內炎 (endovasculitis),暗示可能的自體免疫致病機轉。肺靜脈與小靜脈的纖維化可能類似於肺靜脈閉塞性疾病 (pulmonary veno-occlusive disease) 所觀察到的變化。肺高壓與抗著絲點抗體的存在相關。細支氣管炎主要影響終末與呼吸性細支氣管。除了慢性發炎外,可見細支氣管鱗狀化生 (squamous metaplasia) 與伴管腔縮窄的不定量瘢痕形成。
近期一項涉及共 344 名小兒發病與成人發病局限性硬皮病病人的研究,分別在 27% 與 17% 的病人中偵測到疾病復發。復發在發生於四肢的線狀局限性硬皮病病人中較頻繁,且與發病年齡無關。
局限性硬皮病與系統性硬化症之間的精確關係不確定。由於臨床與病理的重疊,一些作者相信這兩種狀況代表結締組織損傷譜系的兩個極端,類似於盤狀與系統性紅斑性狼瘡之間的關係。確實,病人罕見同時有硬斑病與進行性系統性硬化症(前者通常先於後者);然而,此現象發生得如此罕見,以致大多數人相信此關係純屬巧合。或者,這兩種疾患的特徵可能僅僅代表由相當不同機制所造成的組織損傷的共同表現,類似於可能導致過敏性血管炎 (allergic vasculitis) 組織學外觀的廣泛致病因子範圍。
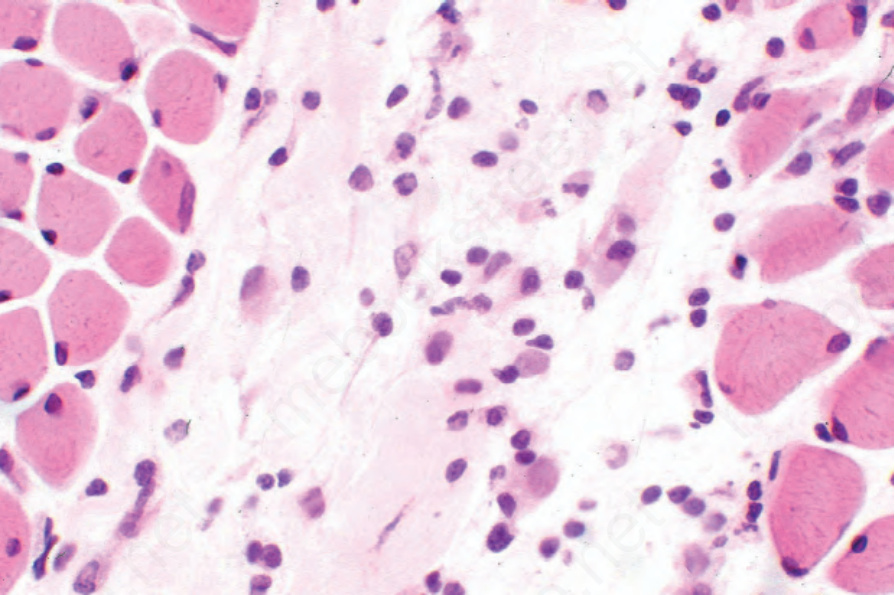
圖 17.101:系統性硬化症 (systemic sclerosis):肌炎,特徵為淋巴組織球浸潤 (lymphohistiocytic infiltrate) 與局灶性骨骼肌再生。
Fig. 17.101 Systemic sclerosis: myositis characterized by a lymphohistiocytic infiltrate and focal skeletal muscle regeneration.
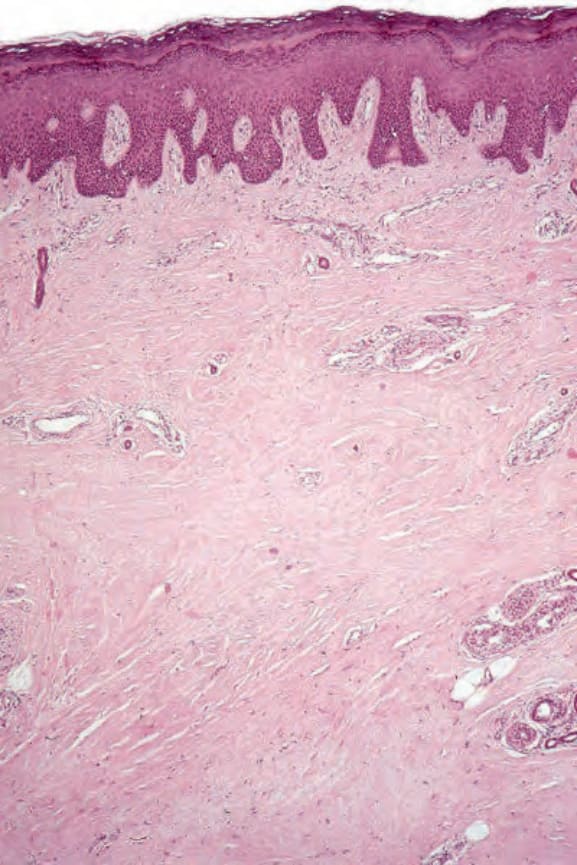
圖 17.96:系統性硬化症 (systemic sclerosis):肢端皮膚的掃描視野,顯示真皮纖維化。此檢體為因嚴重血管侵犯而施行的足部截肢。
Fig. 17.96 Systemic sclerosis: scanning view of acral skin showing dermal fibrosis. The specimen was a foot amputation performed because of severe vascular involvement.
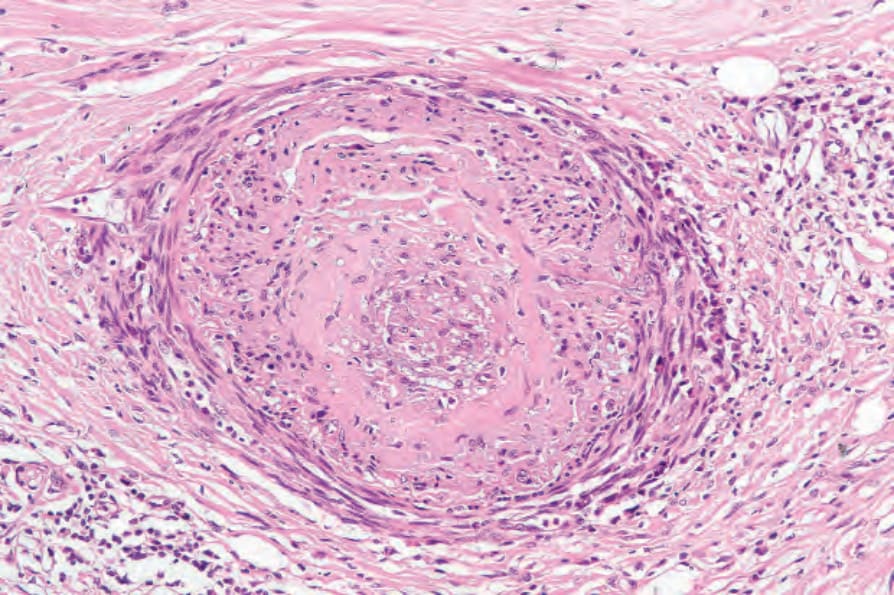
圖 17.98:系統性硬化症 (systemic sclerosis):嚴重的血管侵犯,特徵為內膜纖維化 (intimal fibrosis) 與管腔閉塞。注意周圍的慢性發炎與瘢痕形成。
Fig. 17.98 Systemic sclerosis: severe vascular involvement characterized by intimal fibrosis and obliteration of the lumen. Note the surrounding chronic inflammation and scarring.
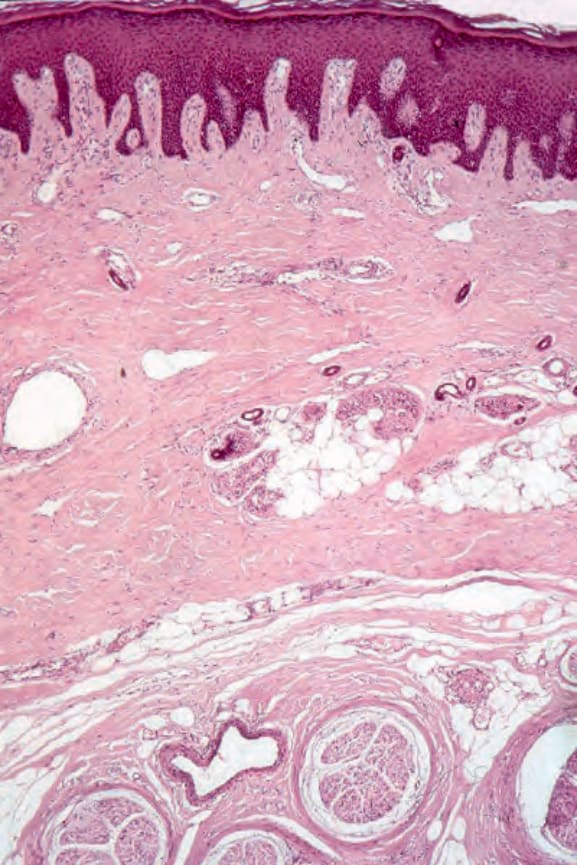
圖 17.99:系統性硬化症 (systemic sclerosis):有戲劇性的神經周圍纖維化 (perineural fibrosis)。
Fig. 17.99 Systemic sclerosis: there is dramatic perineural fibrosis.
最重要的胃腸道病灶是食道平滑肌的萎縮伴纖維化;類似的變化也可能在小腸與大腸中發展。血管肌內膜增生 (vascular myointimal proliferation) 伴管腔狹窄通常也明顯。逆流性食道炎 (reflux esophagitis) 除了慢性發炎變化外,可能顯示糜爛 (erosions) 與潰瘍區。與傳統憩室相反,系統性硬化症的憩室由腸壁的所有層次組成。
臨床特徵 (Clinical features) 局限性硬皮病包含多種狀況,它們可能獨立發生,但常一起出現:
- 斑塊型 (plaque-form)(最常見的變異型),
- 大皰性硬斑病 (bullous morphea),
- 滴狀病灶 (guttate lesions),
- 線狀硬斑病 (linear morphea),包括顏面半側萎縮 (facial hemiatrophy),
- 全身性硬斑病 (generalized morphea),
- 皮下硬皮病 (subcutaneous scleroderma)(深部硬斑病,morphea profunda),
- 兒童致殘性全硬化性硬斑病 (disabling pansclerotic morphea of children)。
早期心肌變化的特徵為肌纖維壞死,伴慢性發炎細胞與組織球浸潤。隨後的纖維化以相等的頻率影響右心室與左心室。主要冠狀動脈在系統性硬化症中顯得正常,但小動脈、內皮與內膜增生伴壁瘢痕形成很常見。
在活躍病灶中,滑膜活檢顯示濃重的表面纖維蛋白沉積。鄰近有慢性滑膜炎 (synovitis),混合淋巴球與漿細胞。類風濕性關節炎所見的伴生發中心形成的淋巴濾泡並非其特徵。隨著慢性化,滑膜瘢痕形成隨之而來。
斑塊型與線狀硬斑病 (Plaque-form and linear morphea) 斑塊型與線狀硬斑病在女性中較常見(3 : 1),且與進行性系統性硬化症相反,常發生於兒童期。線狀硬斑病在高達 20% 的病人於第一個十年結束前發展,並在高達 75% 的病人於第四個十年發展。局限性斑塊發生於生命中稍晚,雖然 75% 的病人在發病時介於 20 與 50 歲之間。