疾病定義與臨床特徵
- 腸病性肢端皮膚炎(acrodermatitis enteropathica)為罕見的體染色體隱性遺傳鋅吸收障礙疾病,主要影響嬰兒,對飲食補充鋅反應顯著。
- 臨床表現為腹瀉、口炎、易怒與生長遲滯,伴隨紅斑、鱗屑與結痂病灶,含水疱、膿疱與糜爛,主要影響四肢、會陰與孔口周圍區域;曾報告明顯大疱病灶。
- 可有非瘢痕性禿髮;其他特徵包括甲失養、傷口癒合延長、膿疱瘡化、身材矮小、精神症狀與畏光;角膜病灶與視力下降亦曾例外報告。
- 病人易發生感染,尤其細菌與真菌,顯示正常鋅濃度對維持免疫系統完整性的重要性。
- 可持續至成年,或罕見於成年首次診斷。
後天性與相關疾病
- 後天型可併發於人工餵養或罕見於母乳餵養嬰兒(足月或早產);因母乳鋅濃度低(源於母血鋅攝入乳房之缺陷);早產兒因腸胃道鋅吸收低及體內鋅儲存低(妊娠最後 10 週由母轉胎)而加重。
- 多種後天性鋅缺乏狀況可出現本病徵象,包括 Crohn 病、酒精性肝硬化、酒精性胰臟炎、腸繞道手術、血液惡性腫瘤化療、神經性厭食、淋巴瘤、生物素缺乏、透析、囊性纖維化、Hartnup 病、必需脂肪酸缺乏、瓜胺酸血症、鳥胺酸轉胺甲醯酶缺乏、全靜脈營養後及乳糜瀉。
- 類似表現亦見於數種胺基酸病與有機酸血症,包括甲基丙二酸血症、丙酸血症、glutaric aciduria type I 與非酮性高甘胺酸血症。
- 變化不僅因鋅缺乏,也因含 isoleucine 等支鏈胺基酸缺乏(由低蛋白飲食誘發);本病亦曾與 HIV 感染相關。
致病機轉
- 病徵源於腸道對鋅吸收不足,機轉涉及鋅轉運蛋白缺陷。
- 早期研究的 SLC30A4 與 ZNT4 基因未顯示相關;後來於染色體 8q24.3 鑑定出致病基因 SLC39A4,編碼富含組胺酸的跨膜蛋白 hZIP4,參與鋅攝取。
- 突變亦影響纖維母細胞的鋅代謝並降低 5′-nucleotidase 活性;迄今已描述超過 30 種不同突變。
- 絕大多數病人帶有 SLC39A4 的同型合子或複合異型合子突變;部分病人無可辨識基因突變,提示其他遺傳因素可能參與。
- 感染機轉與鋅缺乏導致的免疫系統改變相關;鋅對淋巴球、嗜中性球、巨噬細胞、NK 細胞發育與功能及細胞激素產生至關重要,並具抗氧化作用。常見淋巴球減少與胸腺萎縮,源於骨髓 B、T 細胞前驅細胞流失;鋅缺乏誘發糖皮質素媒介的細胞凋亡,致淋巴生成減少。
組織病理特徵
- 組織病理隨病灶演化階段而異。
- 極早期病灶呈細微變化,為局部角化不全與正角化交替。
- 進展時角化不全更明顯且融合,顆粒層減少或消失;上層表皮角質細胞顯著胞質蒼白,並有局部海綿水腫;偶見角化不良細胞。
- 晚期出現胞質空泡化與壞死,可形成表皮內水疱或偶進展為水疱形成;可見角質下膿疱,通常提示次發性感染。
鑑別診斷
- 組織學上與壞死性遊走性紅斑(necrolytic migratory erythema)及糙皮病(pellagra)無法區分。
- 壞死性肢端紅斑(necrolytic acral erythema)亦有極相似組織學特徵,發生於 C 型肝炎病人足背與小腿,病灶為紅斑與乾癬樣斑塊,伴血清與病灶鋅濃度下降。
- 上層表皮角質細胞顯著蒼白亦見於 lactate dehydrogenase M 次單元缺乏,其皮膚表現稱為每年復發性肢端紅斑(annually recurring acroerythema)。
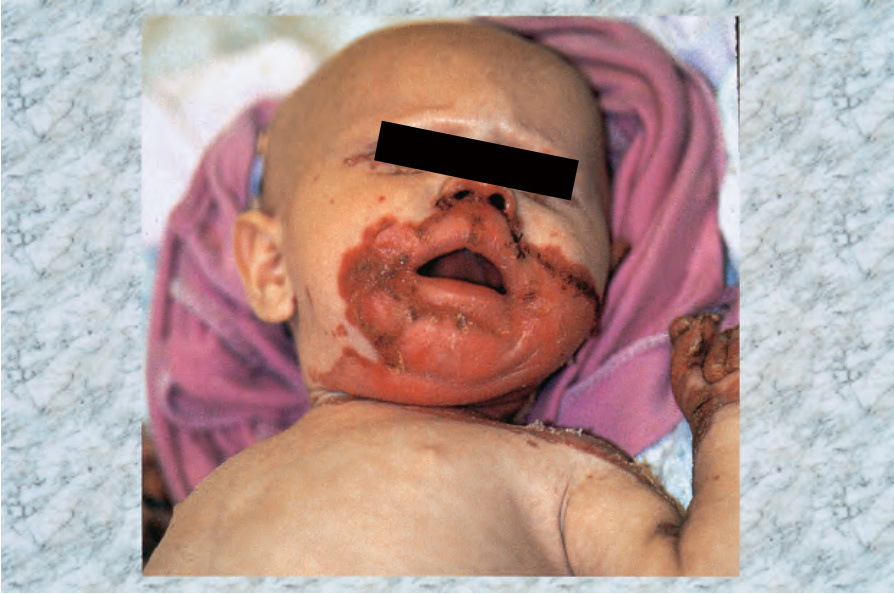
圖 13-199:腸病性肢端皮膚炎:特徵分布的廣泛結痂糜爛。
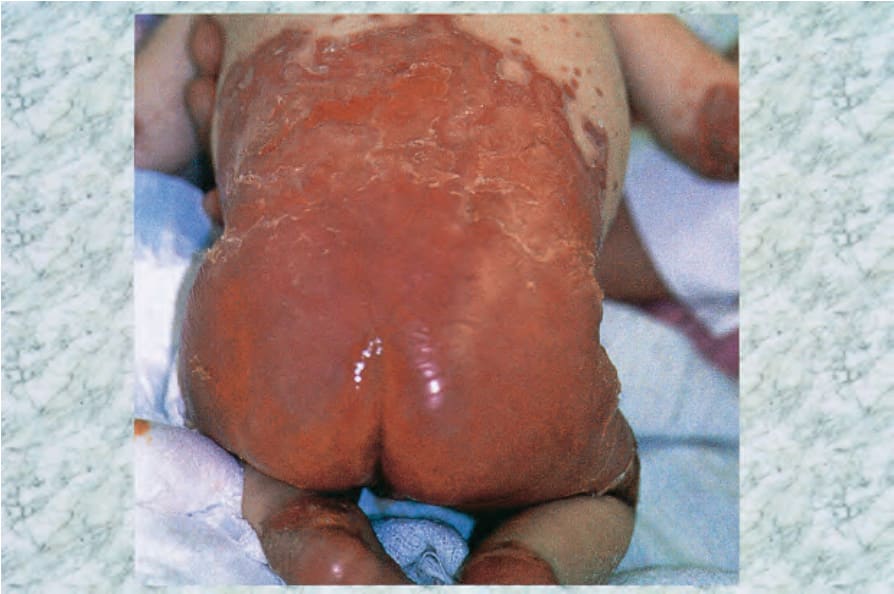
圖 13-200:腸病性肢端皮膚炎:此嬰兒極廣泛侵犯並廣泛糜爛。
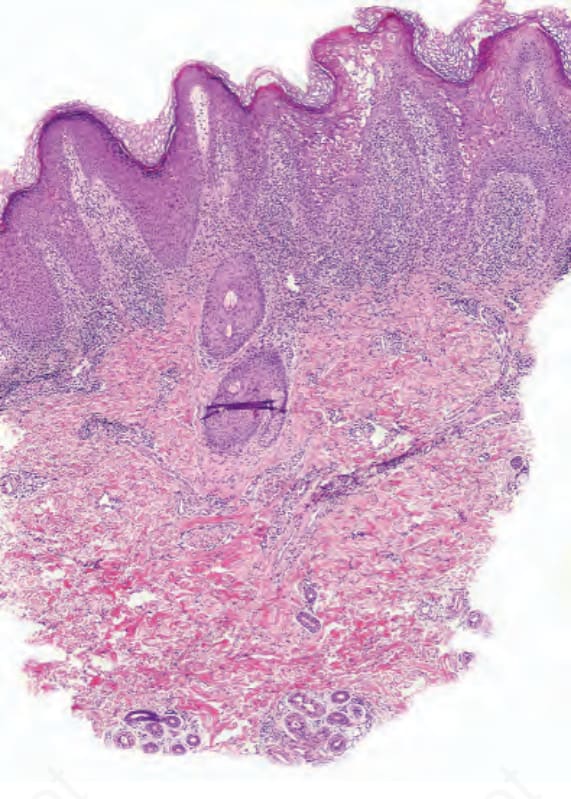
圖 13-201:腸病性肢端皮膚炎:低倍視野顯示角化過度與右側顯著表皮嗜伊紅性。
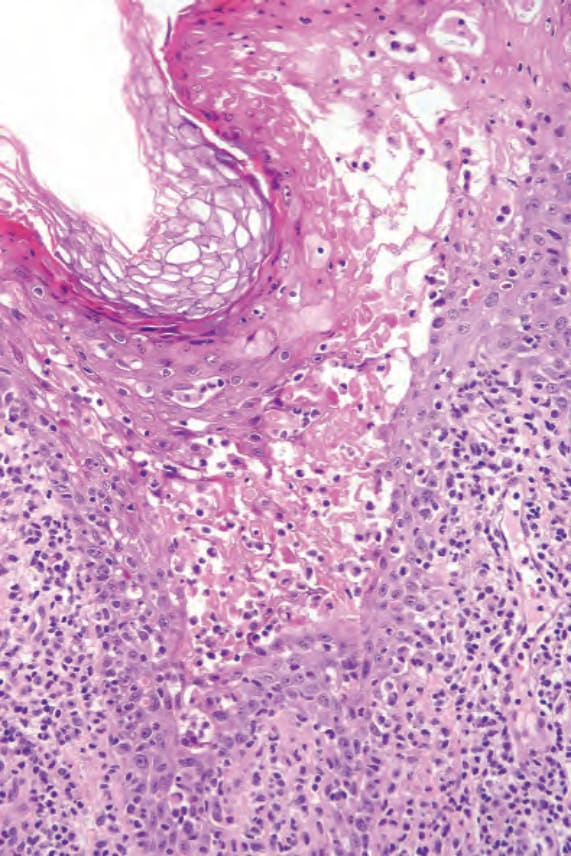
圖 13-202:腸病性肢端皮膚炎:高倍視野可見角質細胞壞死。