疾病定義
- 移植物對抗宿主病 (graft-versus-host disease, GVHD) 為器官移植(多為骨髓移植)後的複雜多系統重大併發症,特別侵犯皮膚、腸道與肝臟。
- 當移植入之具免疫能力 donor T lymphocytes 在受贈者免疫抑制背景下,活化、增殖並對宿主 MHC 組織不相容抗原起反應時發生。
- 同卵 class I HLA 抗原相同時(如兄弟姊妹捐贈),class II HLA 抗原 (HLA-DR、-DP、-DQ) 與 minor histocompatibility antigens 具重要致病意義;這些抗原在 pregraft 放療或化療後表現於宿主上皮細胞,使供者淋巴球免疫反應聚焦於皮膚、肝臟與腸道。
- 為 allogeneic 骨髓移植非常嚴重的併發症,發病率與死亡率極高;亦可見於實質器官移植後、嚴重免疫低下者輸注未照射血品後,或免疫缺陷胎兒接受母體淋巴球經胎盤轉移時。
- 即使 A、B、D loci 完全相同,仍有 35% 病例可發生 GVHD,顯示 minor histocompatibility antigens (miH) 的額外重要性。
- 急性 GVHD 的發生與 HLA disparity、性別不匹配、患者年齡增加及感染相關。
分類
- 傳統依移植後時間分為兩亞群:
- 急性 GVHD:移植後前 3 個月內(最常於第 2–6 週發生)。
- 慢性 GVHD:第 3 個月後。
- 2005 NIH 共識計畫提出新標準分為四群:Classic acute GVHD(移植或捐贈者白血球輸注後 100 天內)、Persistent/recurrent/late onset acute GVHD(移植 100 天後但無慢性症狀)、Classic chronic GVHD、Overlap syndromes(同時具急慢性特徵)。
急性 GVHD (Acute GVHD)
- 發生於 6–90% 接受骨髓移植者;發生率與 HLA mismatch、患者年齡及 conditioning 方案相關。
- 危險因子:性別不匹配(捐贈者為女性、尤其多產,受贈者為男性)、移植前放療/高劑量化療、先前輸血、先前脾切除、病毒感染、免疫抑制不足。
- 臨床傳統分為四期 (stages):
- Stage I:斑丘疹影響達 25% 體表;bilirubin 2–3 mg/dL,腹瀉 >500 mL/day。
- Stage II:斑丘疹紅斑影響 25–50% 體表;bilirubin 3–6 mg/dL,腹瀉 >1000 mL/day。
- Stage III:泛發紅皮症 (erythroderma);bilirubin 6–15 mg/dL,腹瀉 >1500 mL/day。
- Stage IV:toxic epidermal necrolysis;bilirubin ≥15 mg/dL,腹瀉 >1500 mL/day。
- 臨床突發發燒與倦怠,續以臉部紅斑及泛發性 morbilliform 斑丘疹(特徵性侵犯手掌與腳掌);可有黏膜病灶;好發於身體上半部與後頸、耳、肩。
- 嚴重變異型含 erythroderma 或 toxic epidermal necrolysis 樣反應,後者預後差、死亡率很高(50% 以上,尤其未治療者)。
- 臨床與組織學上難以與病毒疾病、cytotoxic/不良藥物反應區別。
慢性 GVHD (Chronic GVHD)
- 發生於 10% 接受 allogeneic 骨髓移植者,及 30–70% 長期存活者;幾乎所有患者有皮膚表現,90% 出現口腔病灶。
- 可 de novo 發生 (30%)、由持續急性 GVHD 漸進 (32%),或在急性緩解後一段靜止期後再發 (36%)。
- 可呈 lichen planus 樣、poikiloderma 樣或 sclerodermatous 反應;亦有多樣早期細微表現(xerosis、ichthyosis、follicular prominence、pityriasiform、eczematous、psoriasiform、annular 病灶、erythroderma 等)。
- 早期病灶常呈典型 lichenoid 外觀,紅色或紫色多角形丘疹,可見 Wickham striae,好發眶周、耳、掌蹠;口腔黏膜呈網狀蕾絲狀白色病灶與潰瘍,常伴 Sjögren syndrome 症狀。
- 晚期典型呈 sclerodermatous,通常於移植後 8–18 個月出現;poikiloderma 樣疹後續硬結、萎縮、硬化,類似 morphea 或 systemic sclerosis。
- 死亡率達 40%,死因含感染、惡病質與肝衰竭;全身表現含慢性肝炎、腹瀉合併吸收不良、bronchiolitis obliterans、周邊壓迫性神經病變與 polymyositis。
- 危險因子:先前急性 GVHD、年齡增加、性別不匹配、使用未去 T 細胞之骨髓。
致病機轉 (Pathogenesis)
- 由 donor T lymphocytes(CD4+ 對 MHC class II、CD8+ 對 class I)與細胞激素(IL-1、TNF-α、IFN-γ、GM-CSF)共同媒介。
- Th1 (產 IL-2、IFN-γ) 促進 GVHD;Th2 (產 IL-4、IL-6、IL-10) 被認為具保護性;B 細胞缺乏;NK 細胞重要性不定。
- 化療/放療後活化的角質細胞產生 TNF-α 與 IL-1 並表現 ICAM-1 與 HLA-DR;內皮細胞表現 E-selectin、整合素、ICAM-1、PECAM-1、VCAM-1 等媒介淋巴球黏附。
- 細胞損傷與死亡來自 cytotoxic T cell(perforin、granzyme B)與經 Fas-Fas ligand 途徑之 apoptosis;血清 TNF-α 升高與 GVHD 相關。
- 部分急性 GVHD 患者 (達 39%) 在真皮-表皮交界與淺層血管周圍有 IgM 與 C3 沉積,提示體液反應可能參與。
- 慢性 GVHD 浸潤以 CD8+ T cells 為主,NK 細胞通常缺乏;TNF-α 與 IL-1 為主要相關細胞激素。
組織病理特徵 (Histopathology)
- 急性病灶以局部或瀰漫性 basal cell hydropic change 為特徵;表皮各層出現 apoptotic/dyskeratotic 角質細胞,鄰近淋巴球 (satellite cell necrosis) 為特徵;可見 cytoid bodies、淋巴球 exocytosis、有時 spongiosis、真皮-表皮交界 microvesiculation。
- 常見毛囊侵犯(hair bulge region),Langerhans cells 數量常減少;血管變化含內皮腫脹脫落、血管周圍淋巴球浸潤。
- 嗜酸性球有時出現,但數量眾多 (≥16/10 HPFs) 提示藥物反應。
- 急性 GVHD 組織學可分四級 (grading),具預後意義(Grade I:basal cell 空泡變性;Grade II:併 spongiosis 與 dyskeratosis;Grade III:subepidermal cleft;Grade IV:表皮完全脫失)。
- Toxic epidermal necrolysis 樣病灶呈嚴重表皮壞死併 subepidermal vesiculation,常見汗腺受侵。
- 慢性 GVHD 組織學典型呈 lichenoid,與 idiopathic lichen planus 高度重疊(hyperkeratosis、hypergranulosis、不規則 acanthosis、basal cell hydropic degeneration、cytoid body、pigmentary incontinence、帶狀 lymphohistiocytic 浸潤);早期常見 satellite cell necrosis,浸潤有時含漿細胞與嗜酸性球,密度較低。
- 晚期慢性 GVHD 呈表皮萎縮、ridge pattern 消失、淺深層真皮疤痕化、附屬器消失,呈 sclerodermoid 樣(含 eosinophilic fasciitis、panniculitis、morphea 樣、lichen sclerosus)。
鑑別診斷 (Differential Diagnosis)
- 急性 GVHD 特徵可由 cytotoxic drugs(如 cyclophosphamide)與放療重現;亦須與病毒感染、不良藥物反應鑑別;顯著嗜酸性球較傾向藥物反應,但組織學上未必能區別。
- 急性 GVHD 可與 erythema multiforme、嚴重者與 toxic epidermal necrolysis 難分;GVHD 少數病人角質層有 bile pigment 沉積(erythema multiforme 無)。
- 淋巴球恢復疹 (eruption of lymphocyte recovery) 與急性 GVHD 無法區別;engraftment syndrome 亦進入鑑別(移植後 10–14 天,發燒、肝炎、腸道症狀與紅斑斑丘疹)。
- 早期慢性 GVHD 可與 lichen planus 無法區別,但真皮浸潤較不明顯、有時含漿細胞與嗜酸性球;satellite cell necrosis 可為診斷指標。
- 無單一組織學特徵可作為 GVHD 之 pathognomonic 病徵,臨床相關不可或缺。
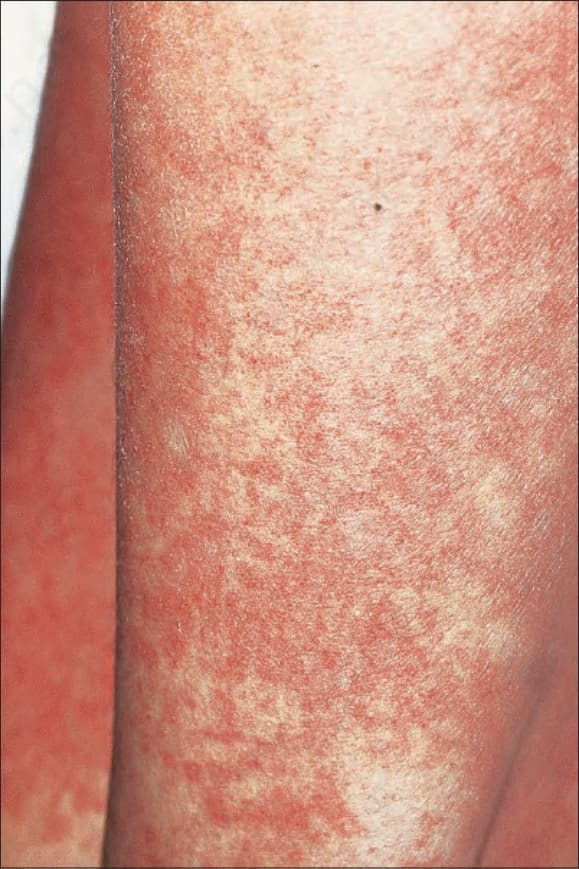
圖 7-106:Acute GVHD 胸部與手臂呈廣泛黃斑紅斑併細微毛細血管擴張與輕度脫屑。
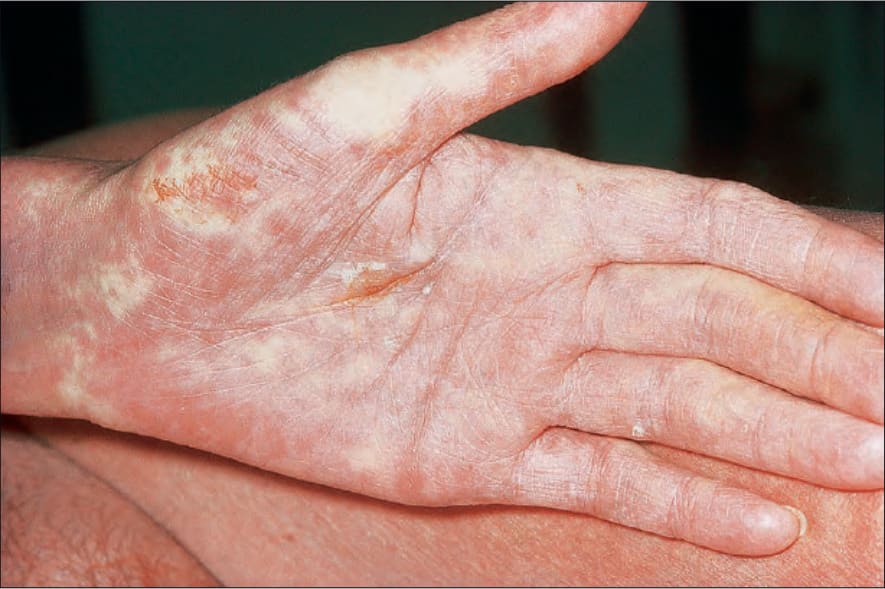
圖 7-107:Acute GVHD 特徵性鮮明掌部紅斑。
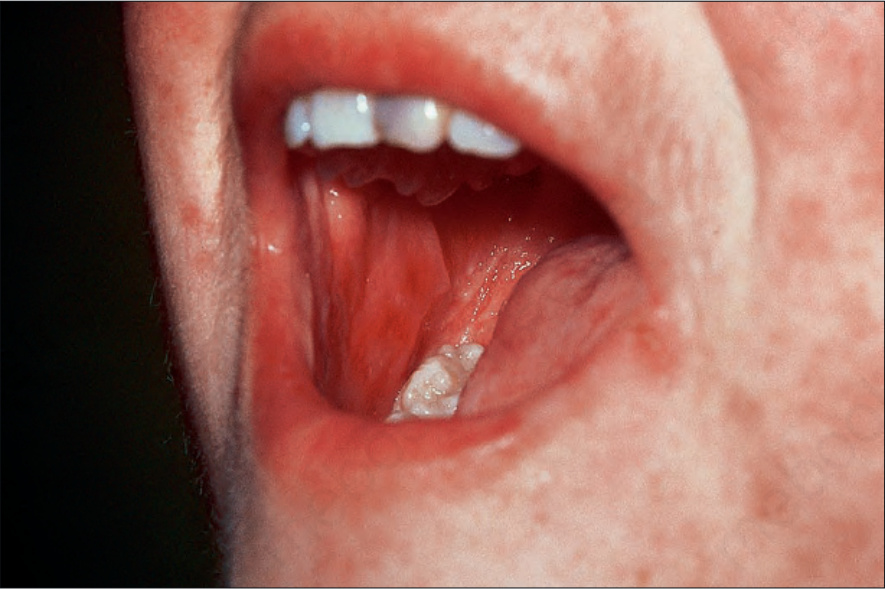
圖 7-108:Acute GVHD 頰黏膜糜爛。
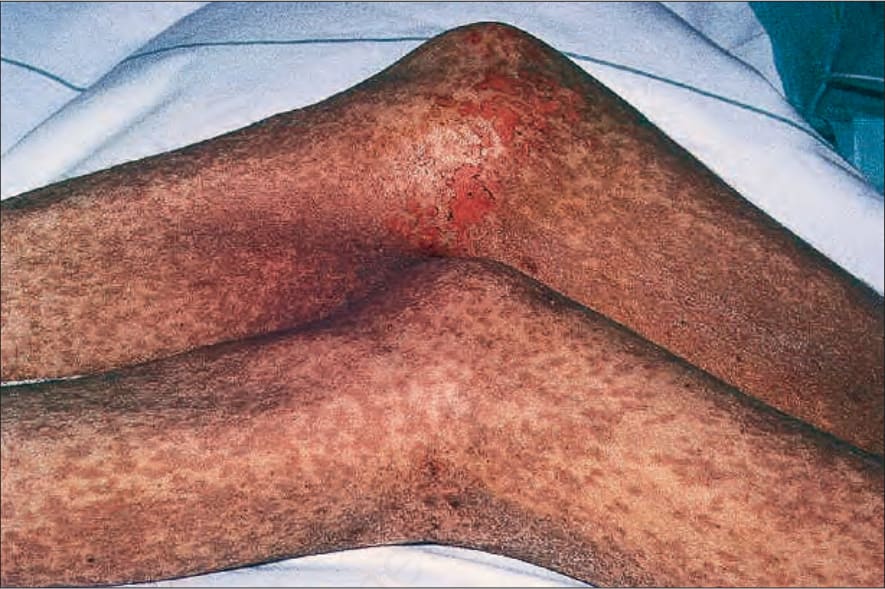
圖 7-109:早期 chronic GVHD 廣泛近融合性色素沉著 lichenoid 丘疹併表皮糜爛。
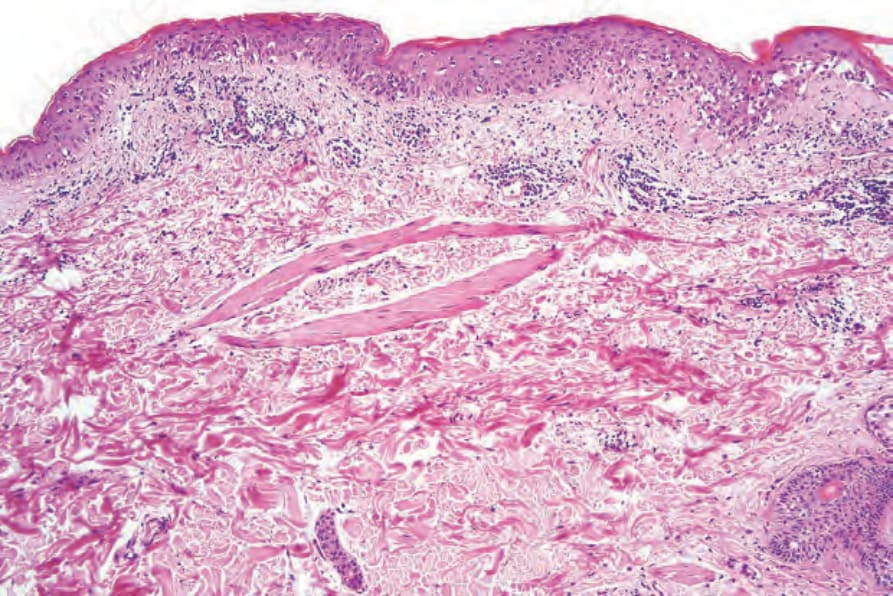
圖 7-116:Acute GVHD 演變中病灶顯示 basal cell hydropic degeneration 與散在 apoptotic 角質細胞。
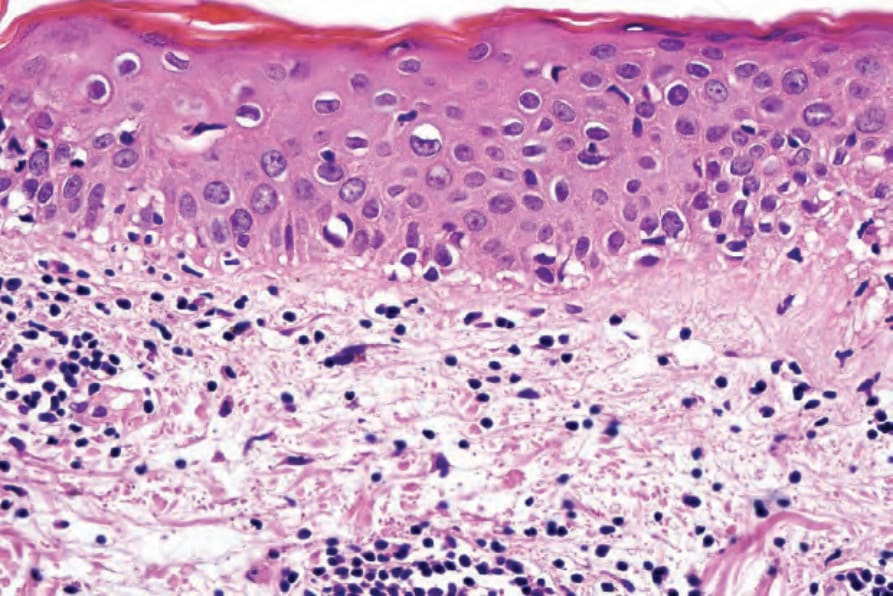
圖 7-117:Acute GVHD 高倍視野顯示 basal cell liquefactive degeneration。
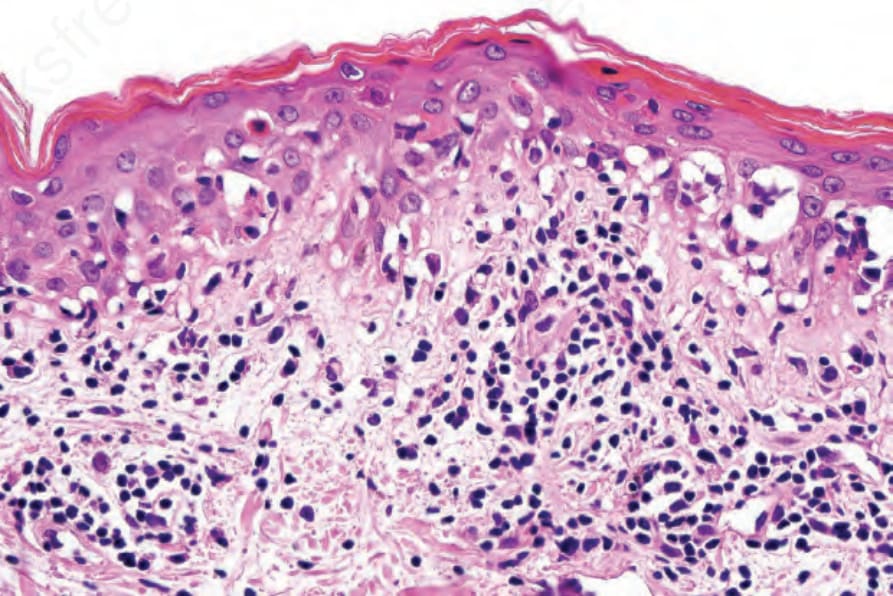
圖 7-118:Acute GVHD 高倍視野顯示 parakeratosis、basal cell hydropic degeneration 與 apoptosis。
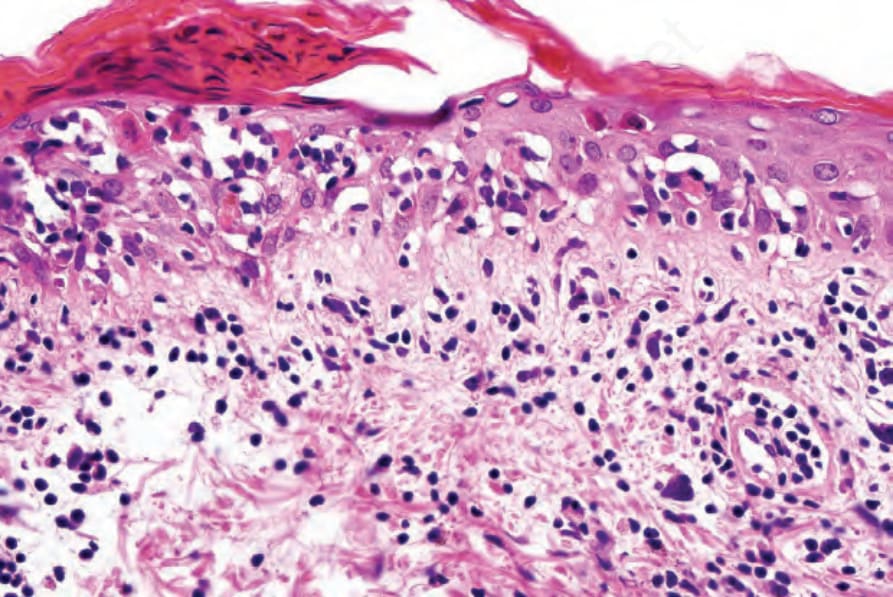
圖 7-119:Acute GVHD 高倍視野顯示 parakeratosis、apoptosis 與 satellite cell necrosis。
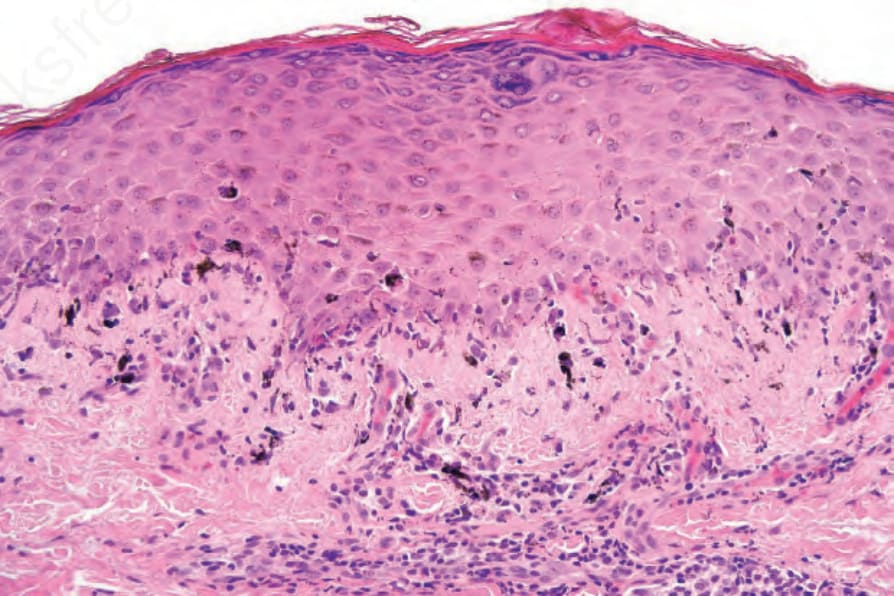
圖 7-120:早期 chronic GVHD 之 hyperkeratosis、hypergranulosis、不規則 acanthosis 與 basal cell hydropic degeneration,與 idiopathic lichen planus 無法區別。
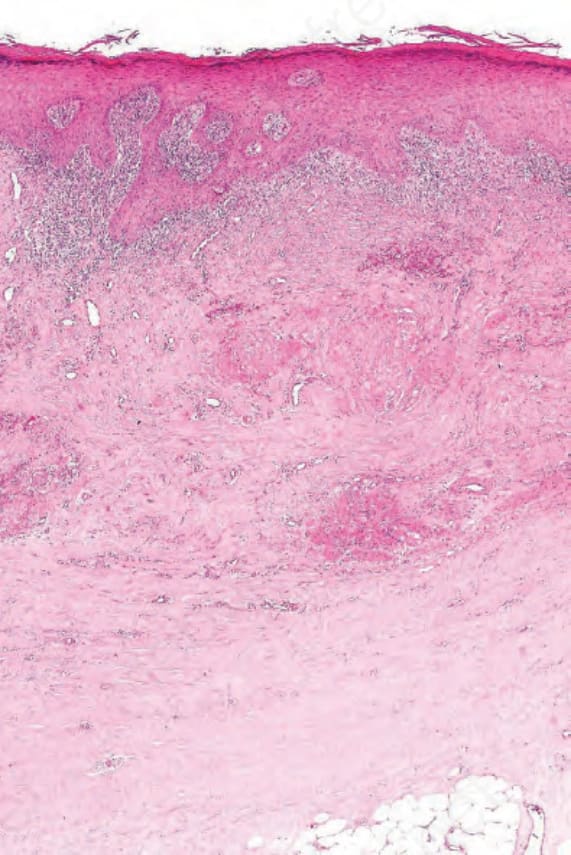
圖 7-121:晚期 chronic GVHD 真皮緻密纖維化併皮下脂肪受牽連,附屬器消失,類似 scleroderma。

表 7-1:Acute graft-versus-host disease 之分級。