Graft-versus-host disease
Graft-versus-host disease
Clinical features GVHD represents a complex multisystem major complication of organ transplantation, usually bone marrow, that particularly affects the skin, intestine, and liver. It develops when transplanted immunocompetent donor T lymphocytes are activated, proliferate, and respond to foreign host major histocompatibility complex (MHC)-histoincompatible antigens in a background of recipient immunosuppression.1–9 In the context of identical class I HLA antigens, as may be seen in sibling donors, class II HLA antigens (HLA-DR, -DP, and -DQ) and minor histocompatibility antigens are of major pathogenetic significance.1,2 These latter HLA antigens are expressed on host epithelial cells following pregraft irradiation or chemotherapy, thereby focusing the donor lymphocyte immune response on the skin, liver, and intestinal tract.1,10,11
GVHD is a very serious complication of allogeneic bone marrow transplantation and morbidity, and mortality is very high.12 It may also follow solid organ transplantation, develop in severely immunodepressed patients after transfusion of nonirradiated blood or blood products, or complicate transplacental transfer of maternal lymphocytes into an immunodeficient fetus.13–15
affected. Hematological manifestations include thrombocytopenia, aplastic anemia, pancytopenia, myelodysplasia, and acute myeloid leukemia.8,9,11–13
The grave outlook of dyskeratosis congenita relates particularly to the development of infections complicating aplastic anemia, malignancy, and pulmonary complications.2,9–11,14
The clinical features of GVHD develop as a consequence of donor T lymphocyte-mediated reactions to host tissues. Successful bone marrow transplantation is dependent on the compatibility of the ABO system blood groups and histocompatibility antigens (HLA). The D locus (HLA class II) is of particular importance; successful transplantation has occurred in the presence of identical D loci with dissimilarities at the A and B loci. However, the development of GVHD is not totally dependent on HLA incompatibility as it can develop in 35% of cases with identical A, B, and D loci, suggesting the additional importance of the minor histocompatibility antigens (miH).2,16
The clinical features of this disease are most severe in males with the X-linked variant. There is considerable variation in autosomal variants, and in some of these patients symptoms may be very mild, allowing a normal life expectancy.2
Pathogenesis and histologic features Dykeratosis congenita is characterized by mutations in genes involved in telomere function, with the affected gene depending on the mode of inheritance.6 X-linked recessive dyskeratosis congenita is due to mutations of the DKC1 gene, which has been mapped to Xq28.15 The mutations, which are predominantly missense, result in single amino acid substitutions in dyskerin, a nucleolar protein believed to be responsible for site-specific pseudouridylation of ribosomal RNA. It is also associated with mutations in the ACD, CTC1, DKC1, NHP2, NOP10, PARN, RTEL1, TERC, TERT,
Development of acute GVHD appears to be a consequence of HLA disparity, sex mismatch, increasing patient age, and the presence of infection.7 While the skin is a major target organ in GVHD and one of the first involved organs, the liver and gastrointestinal tract are also affected.9,17 Manifestations include malaise, nausea and vomiting, diarrhea, malabsorption, and abnormal liver function. Additionally, patients with GVHD have an increased risk of opportunistic infections, which are an important cause of morbidity and mortality.
Historically, GVHD was conventionally subdivided into two subgroups by time after transplantation:
• Acute GVHD occurs within the first 3 months following transplantation (most often presenting between 2 and 6 weeks).2,17–19
• Chronic GVHD presents after the third month. However, changing transplantation practices have resulted in delayed and even atypical presentations of GVHD. In 2005, the National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in
274 Lichenoid and interface dermatitis
Chronic Graft-versus-Host Disease proposed new criteria to standardize the diagnosis of chronic GVHD and also account for these new GVHD presentations, dividing it into four groups17,19–21:
• Classic acute GVHD: acute GVHD presenting within 100 days after hematopoietic stem cell transplant or donor leukocyte infusion,
• Persistent, recurrent, or late onset acute GVHD: acute GVHD occurring more than 100 days after transplantation without chronic GVHD symptoms,
• Classic chronic GVHD: chronic GVHD without features of acute GHVD regardless of timing from transplantation,
• Overlap syndromes: both acute and chronic GVHD features present regardless of timing from transplantation. The classical features of acute and chronic GVHD are presented below. The other two categories show similar features and are defined by the clinical context in which they occur, that is, their timing relative to transplantation.
More severe variants include erythroderma or even a toxic epidermal necrolysis-like reaction. The latter has a poor prognosis and may be a manifestation of a drug reaction or represent a true component of acute GVHD. It usually affects a large surface area, shows mucosal involvement, and is associated with severe liver and gastrointestinal lesions.23–25 Mortality is very high (50% and higher, especially if untreated), related to the effects of therapy in addition to the lesions themselves.12,26 In the event of survival of acute GVHD, the rash may resolve completely or merge into the features of chronic GVHD. It is often difficult on clinical grounds (and histologically) to differentiate between acute GVHD, viral disorders, and cytotoxic/ adverse drug reactions.
Acute GVHD Acute GVHD develops in between 6% and 90% of patients who undergo bone marrow transplantation.22,23 The incidence relates particularly to HLA mismatch, the age of the patient, and the conditioning regimen protocols used.1,2 Additional risk factors of importance include sex mismatch, i.e., when the donor is a female (particularly if multiparous) and the recipient is male, use of radiation and/or high dosage chemotherapy prior to transplantation, prior blood transfusions, prior splenectomy, viral infections, and inadequate immunosuppression.2
The clinical manifestations of acute GVHD are traditionally divided into four stages1,2,17,18,23:
• Stage I: Maculopapular eruption affecting up to 25% of surface area. Bilirubin levels of 2–3 mg/dL and diarrhea in excess of 500 mL/day.
• Stage II: Maculopapular erythema affecting 25–50% of surface area. Bilirubin levels of 3–6 mg/dL and diarrhea in excess of 1000 mL/day.
• Stage III: Generalized erythroderma. Bilirubin levels of 6–15 mg/dL and diarrhea in excess of 1500 mL/day.
• Stage IV: Toxic epidermal necrolysis. Bilirubin levels of 15 mg/dL or more and diarrhea exceeding 1500 mL/day.
It presents with the sudden onset of fever and malaise, which are rapidly followed by cutaneous signs including facial erythema and a generalized morbilliform, maculopapular rash characteristically affecting the palms and soles (Figs 7.106 and 7.107). Mucosal lesions may also be a feature (Fig. 7.108). The skin lesions particularly affect the upper half of the body and the back of the neck; ears and shoulders are sites of predilection.1,7,18,19,23 Lichen planus-like features may sometimes supervene. Additional cutaneous lesions include purpura, petechiae, desquamation, and a folliculitis-like appearance.7,19,23
Chronic GVHD Chronic GVHD develops in 10% of all patients undergoing allogeneic bone marrow transplantation and in 30–70% of all long-term survivors.27,28 Systems involved include the skin, eyes, mouth and esophagus, liver, genitalia, muscle, and peripheral and central nervous systems.7 Virtually all chronic GVHD patients exhibit skin manifestations, and 90% develop oral lesions.2,28,29 Some develop chronic GVHD de novo (30%); others show a gradual progression of continuous acute GVHD into the chronic variant (32%).2 Occasionally, chronic GVHD may follow a period of resolution of acute GVHD, after an interval of quiescence (36%).2 Chronic GVHD can occur as a lichen planus-like eruption and/or show features of a poikilodermatous or sclerodermatous reaction.30–32 A discoid lupus erythematosus-like reaction is rare. Polymyositis and fasciitis have also been described.33–38 In addition, a variety of presentations have been reported which can be subtle, especially in the early phase. These include xerosis, ichthyosis, follicular prominence, pityriasiform, eczematous, psoriasiform lesions, annular lesions similar to urticaria or erthyema annulare centrifigum, a morbilliform papulosquamous rash, and even erythroderma.20 Risk factors for developing chronic GVHD include prior episode of acute GVHD, increasing age, sex mismatch, i.e., when the donor is a female (particularly if multiparous) and the recipient is male, and use of non-T-cell depleted bone marrow.2,39
275 Interface dermatoses
Chronic GVHD has a mortality of up to 40%. Causes of death include infection, cachexia, and liver failure.2
Systemic features include chronic hepatitis, diarrhea with malabsorption, bronchiolitis obliterans, peripheral entrapment neuropathy, and polymyositis.2 Opportunistic infections are also of major importance.
Pathogenesis and histologic features GVHD is mediated by the combined effects of donor T lymphocytes (CD4+ T cells responding to MHC class II antigens and CD8+ T cells to class I antigens) and cytokines including IL-1, TNF-α, IFN-γ, and GM-CSF.1,2,11,45–53 The development of acute GVHD depends on a complex interplay between
Although early in chronic GVHD the lesions are typically lichenoid and later sclerodermatous, in some patients these features may appear simultaneously.2 UV irradiation, trauma, and infection with herpes zoster virus or Borrelia can precipitate chronic GVHD.2
The early chronic GVHD lesion commonly has a classic lichenoid appearance with typical erythematous or violaceous polygonal papules sometimes showing Wickham striae (Fig. 7.109). The periorbital region, ears, palms, and soles are sites of predilection.2 Oral mucosal lesions include typical netlike lacy white lesions, and ulcerated areas may also develop (Figs 7.110–7.112). The cheeks, tongue, palate, and lips are sites of predilection.2 Symptoms of Sjögren syndrome are also often present. Onycholysis and cicatricial alopecia may be features. The rash is sometimes less typical, appearing as a desquamative active dermatitis or as follicular hyperkeratosis. As mentioned above, the findings can sometimes be subtle, such as xerosis.20
The late phase of chronic GVHD is typically sclerodermatous and usually presents 8–18 months after transplantation (Figs 7.113–7.115). The development of a poikilodermatous rash is followed by induration, atrophy, and sclerosis.23,30 The resultant features resemble morphea or systemic sclerosis; chronic ulceration, particularly involving pressure points, can be an unpleasant complication. Blisters may occasionally develop.23,30,33 The development of oral and cutaneous squamous cell carcinoma has occasionally been associated with chronic GVHD.40–44
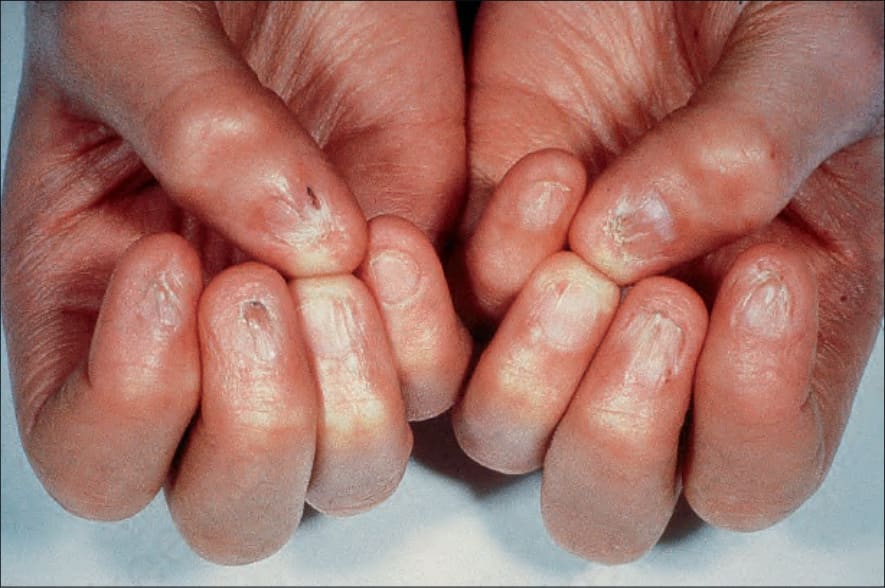
Fig. 7.105 Dyskeratosis congenita: there is dystrophy of the nails with marked atrophy of the surrounding skin. By courtesy of D. Atherton, MD, Institute of Dermatology and Children’s Hospital at Great Ormond Street, London, UK.
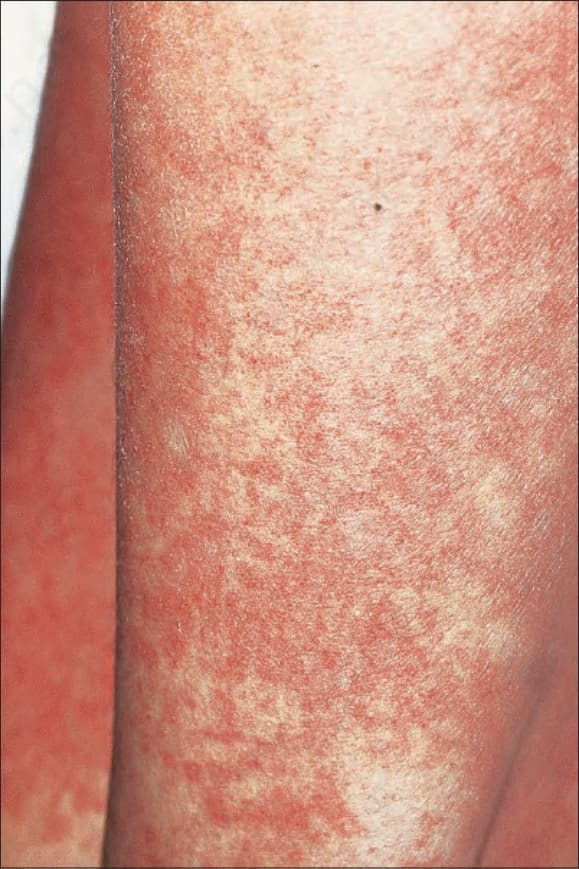
Fig. 7.106 Acute graft-versus-host disease: chest and arm showing widespread macular erythema with fine telangiectasia and mild scaling. By courtesy of R. Touraine, MD, Hôpital Henri Mondor, Paris, France.
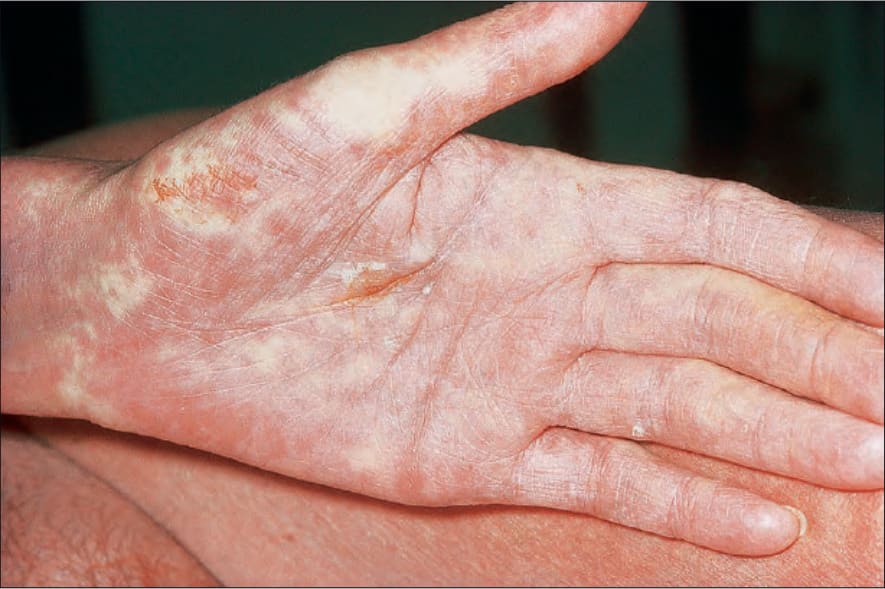
Fig. 7.107 Acute graft-versus-host disease: this vivid palmar erythema is characteristic. By courtesy of R. Touraine, MD, Hôpital Henri Mondor, Paris, France.
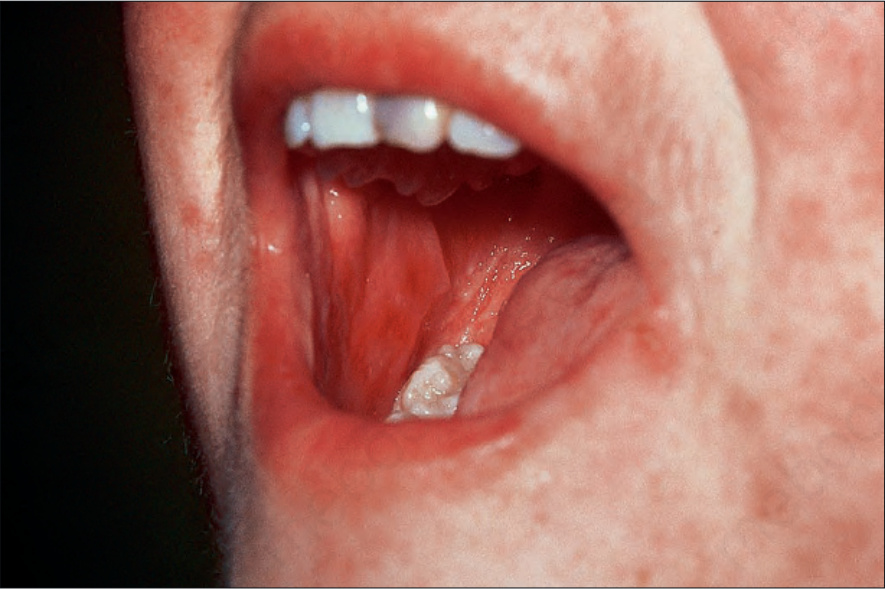
Fig. 7.108 Acute graft-versus-host disease: note the erosions on the buccal mucosa. By courtesy of R. Touraine, MD, Hôpital Henri Mondor, Paris, France.
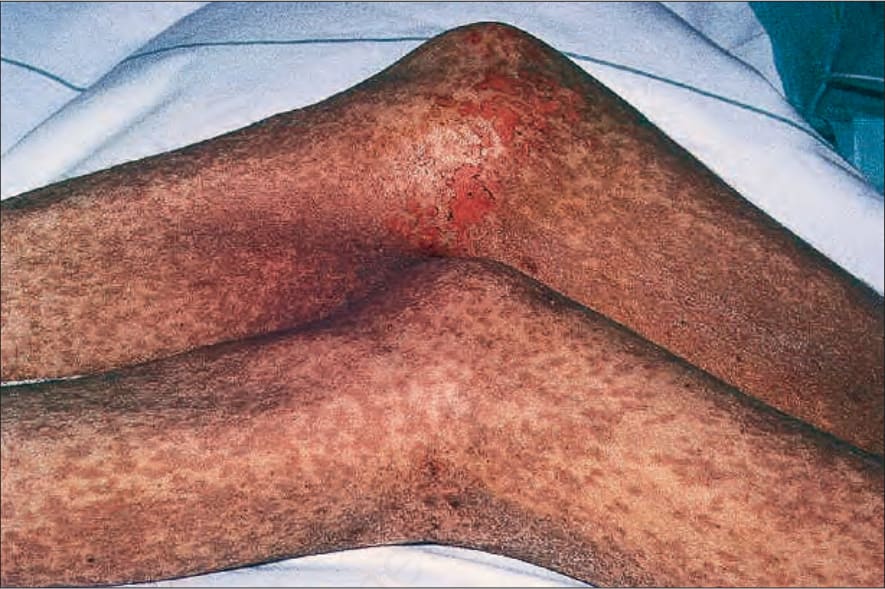
Fig. 7.109 Early chronic graft-versus-host disease: there are widespread, almost confluent hyperpigmented lichenoid papules. Associated erosion of the epidermis gives an appearance similar to toxic epidermal necrolysis (Lyell syndrome). By courtesy of R. Touraine, MD, Hôpital Henri Mondor, Paris, France.
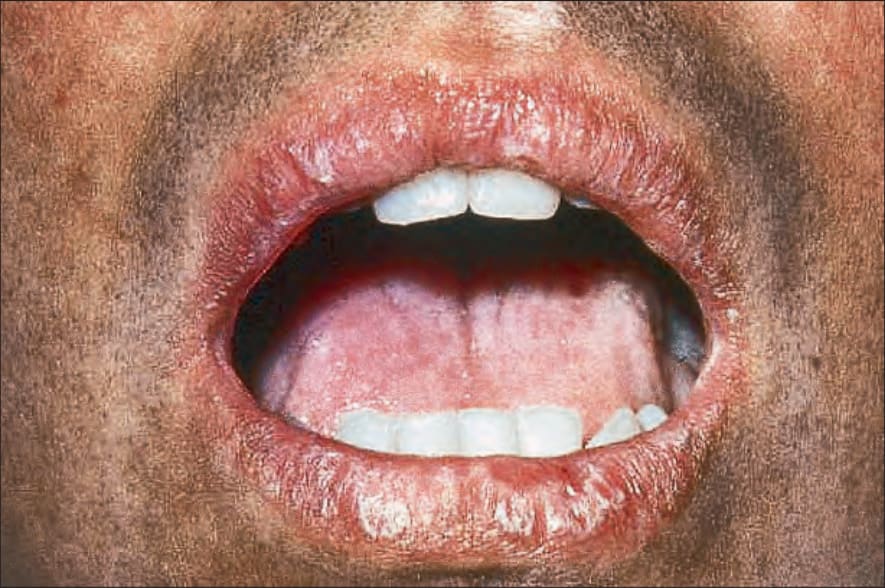
Fig. 7.110 Early chronic graft-versus-host disease: there are diffuse widespread lichenoid changes of the lips. By courtesy of R. Touraine, MD, Hôpital Henri Mondor, Paris, France.
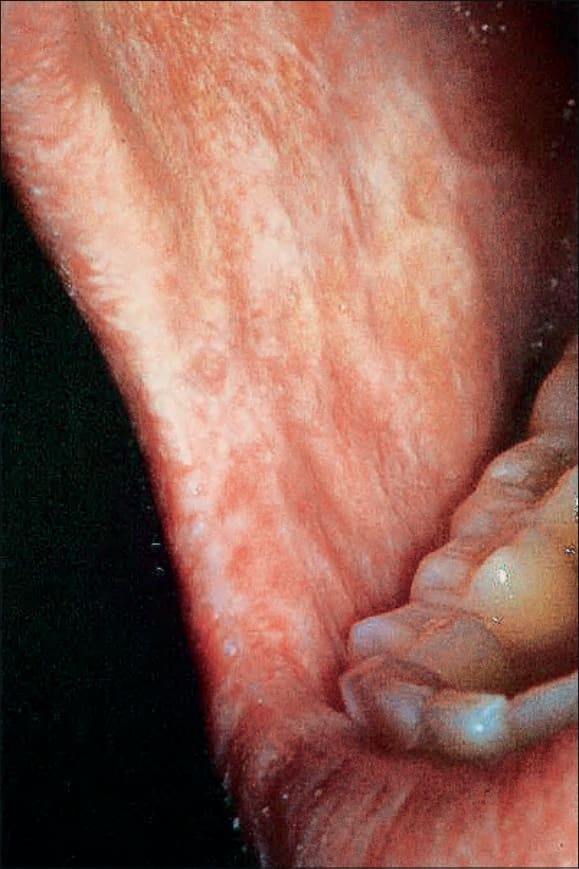
Fig. 7.111 Early chronic graft-versus-host disease: florid reticulate white striae on the buccal mucosa are evident. By courtesy of R. Touraine, MD, Hôpital Henri Mondor, Paris, France.
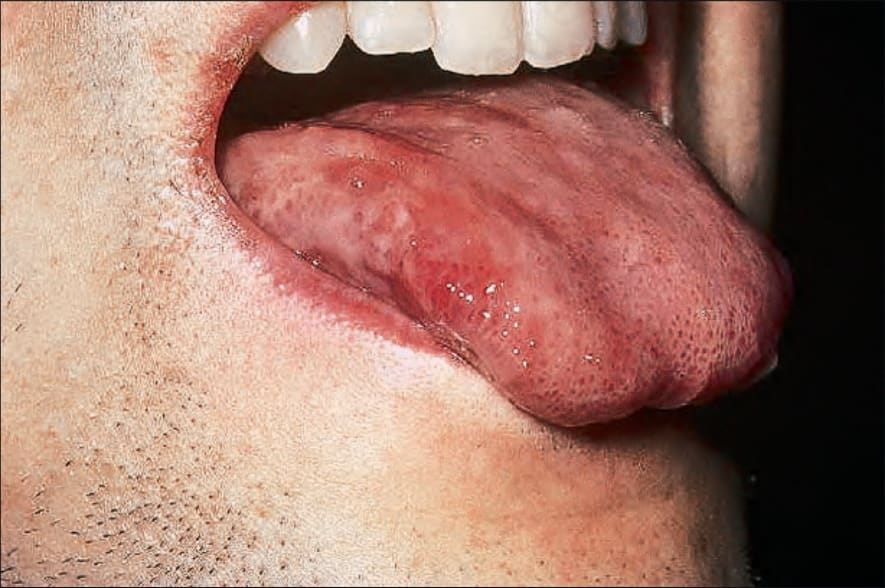
Fig. 7.112 Early chronic graft-versus-host disease: there are erosive changes on the tongue. By courtesy of R. Touraine, MD, Hôpital Henri Mondor, Paris, France.
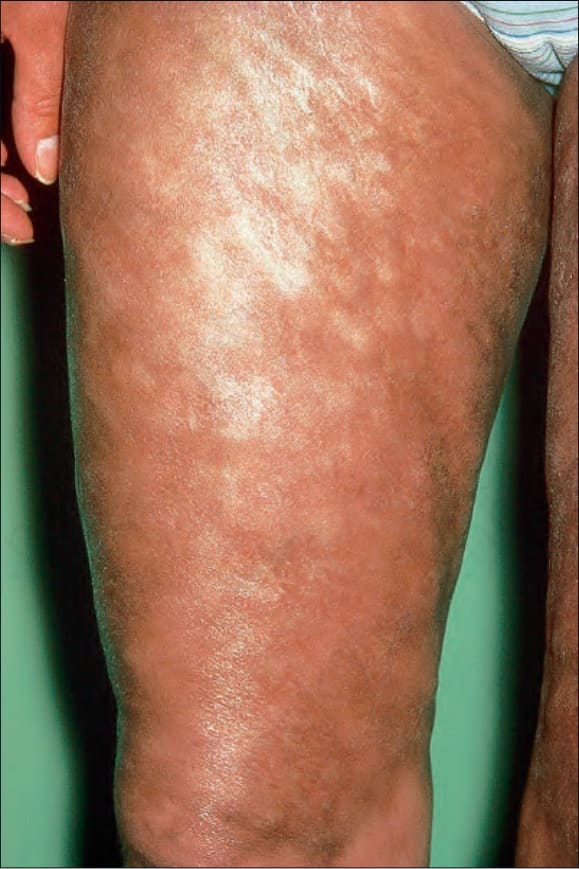
Fig. 7.113 Late chronic graft-versus-host disease: note the grossly hyperpigmented sclerotic limb. By courtesy of R. Touraine, MD, Hôpital Henri Mondor, Paris, France.
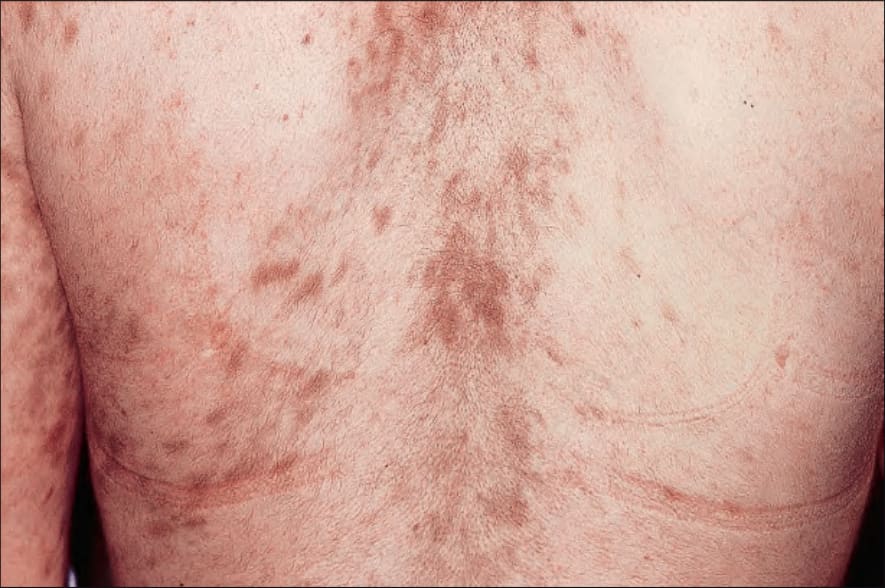
Fig. 7.114 Late chronic graft-versus-host disease: hyperpigmented sclerotic plaques are present on the back. By courtesy of R. Touraine, MD, Hôpital Henri Mondor, Paris, France.
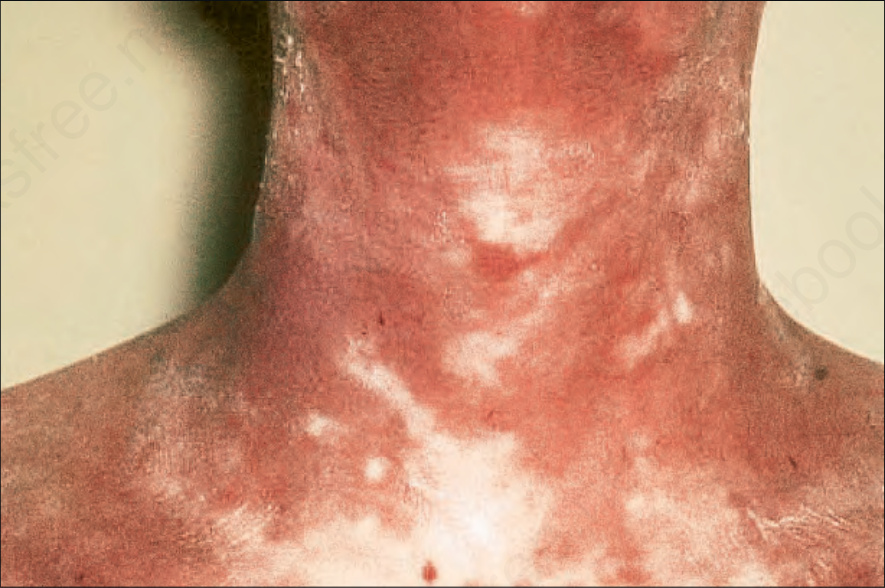
Fig. 7.115 Late chronic graft-versus-host disease: there is mottled hypo- and hyperpigmentation with gross atrophy and scaling. By courtesy of R. Touraine, MD, Hôpital Henri Mondor, Paris, France.
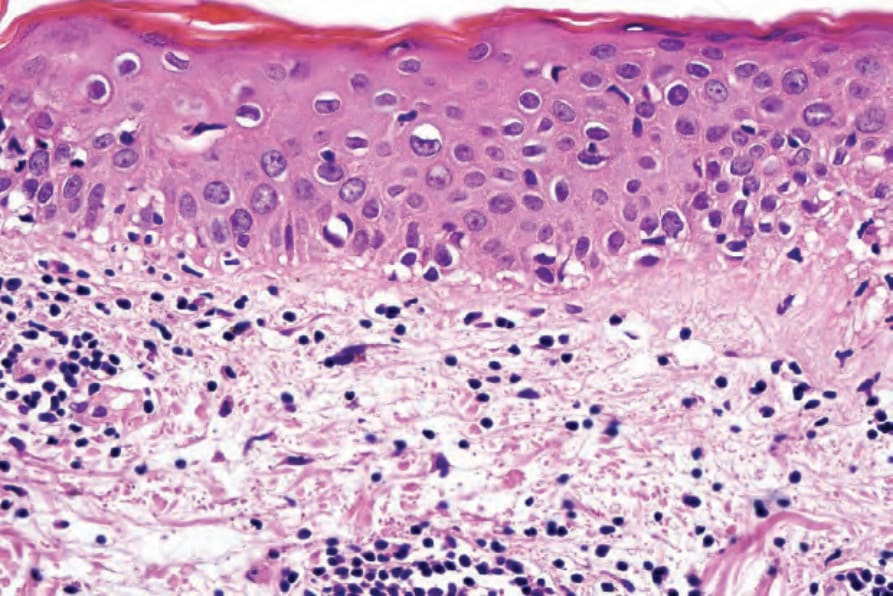
Fig. 7.117 Acute graft-versus-host disease: high-power view showing basal cell liquefactive degeneration. Diagnosis is entirely dependent on the clinical history.
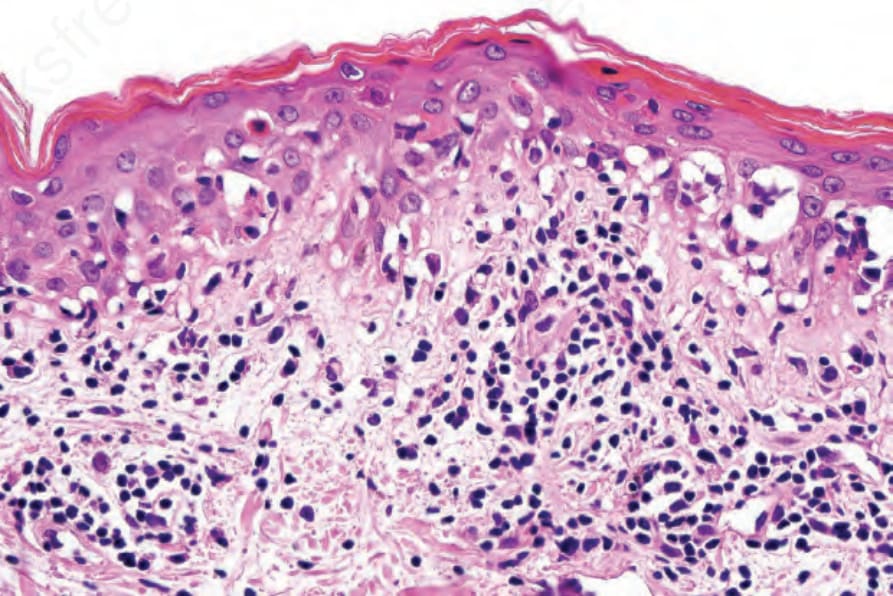
Fig. 7.118 Acute graft-versus-host disease: high-power view lesion showing parakeratosis, basal cell hydropic degeneration, and apoptosis.
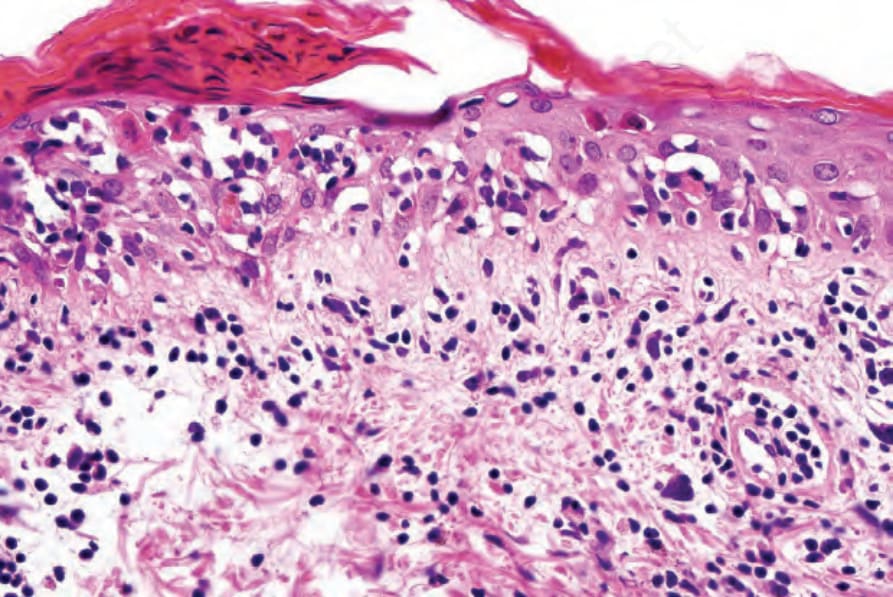
Fig. 7.119 Acute graft-versus-host disease: high-power view showing parakeratosis, apoptosis, and satellite cell necrosis.
276 Lichenoid and interface dermatitis
E-selectin, α4β1 integrin, αLβ2 integrin, ICAM-1, platelet endothelial cell adhesion molecule-1 (PECAM-1), and vascular cell adhesion molecule-1 (VCAM-1), which mediate lymphocyte adhesion to the endothelium and facilitate recognition, activation, and response to MHC molecules.1,57-59 The mechanisms of cell injury and death result from both cytotoxic T cell and possibly NK cell-mediated cytotoxic effects and the actions of cytokines. The former includes cytolytic actions mediated by perforin and granzyme B, and apoptosis through the Fas-Fas ligand pathway.60,61 IL-1, IL-2, IL-6, and TNF-α are thought to be of particular importance in mediating cytotoxicity.1 Raised serum TNF-α correlates with GVHD, and antibodies to TNF-α or its receptor protect against the disease.1,62–64 Recently, regulatory T cells (Treg) have been postulated to play a role in GVHD. Treg are decreased in patients with GVHD, and it appears that they may inhibit tissue infiltration of CD8+ T cells into the affected skin.17,65 Studies suggest a role for B cells in GVHD, with a possible role for targeted therapy.66
host immunosuppression, tissue damage as a result of pregraft induction therapy, and donor lymphocyte proliferation and activation with consequent injury and death of susceptible host tissues.1
The lymphocytes may be of CD4+ or CD8+ immunophenotype, and commonly there is an admixture. Both Th1 and Th2 CD4+ subtypes are represented. The former produce IL-2 and IFN-γ and are thought to promote GVHD; the latter produce IL-4, IL-6, and IL-10 and are believed to be protective, although this has been contested.54 Natural killer (NK) cells may also be of importance although their presence appears to be variable.55 B cells are absent. Activated keratinocytes following induction chemotherapy or irradiation produce TNF-α and IL-1 and express ICAM-1 and HLA-DR.56 This may result in increased recognition of histoincompatible MHC antigens by donor T cells.1 The superficial dermal endothelial cells express
Deposition of IgM and C3 at the dermal–epidermal junction and around the superficial vasculature in up to 39% of patients with acute GVHD suggests that humoral responses may play a role in the pathogenesis of GVHD.56,67
277 Interface dermatoses
The development of chronic GVHD is dependent on a variety of factors including, antihost tissue activity of donor T cells and the development of autoimmunity.2,68 The infiltrate consists predominantly of CD8+ T cells; NK cells are usually absent.2 As with acute GVHD, TNF-α and IL-1 are the major cytokines implicated.2
of GVHD is broad, and no finding can be considered pathognomonic for GVHD.12,19
The toxic epidermal necrolysis-like lesions are characterized by severe epidermal necrosis in association with subepidermal vesiculation. Evidence of sweat gland involvement is commonly present.76,77 Keratinous plugging of the acrosyringium may therefore be seen, and the excretory ducts often show cytopathic-degenerative and proliferative changes.77 The former comprises basal cell hydropic degeneration, lymphocytic infiltration, and apoptosis. Follicular involvement is a not uncommon additional manifestation.78 The histologic features of acute GVHD may be subdivided into four stages, which have prognostic significance (Table 7.1).78–80
The acute lesion of GVHD is characterized by focal or diffuse basal cell hydropic change (Figs 7.116–7.119).69 Apoptotic and dyskeratotic keratinocytes, at all levels of the epidermis and associated with adjacent lymphocytes (satellite cell necrosis), are characteristic.17,70,71 Isolated cytoid bodies are also frequently evident. Lymphocytic exocytosis is invariably present, and spongiosis is sometimes a feature. Microvesiculation at the dermal– epidermal junction occasionally occurs. Follicular involvement is a common feature, and the hair bulge region is typically affected.71,72 Langerhans cells are often reduced in number. Vascular changes include endothelial cell swelling with sloughing, and intimal and perivascular lymphocytic infiltration. Blood vessel proliferation has also been described. Perivascular edema and nuclear dust may additionally be present, and mast cells are also conspicuous.73,74 Eosinophils are sometimes present and this finding does not necessarily indicate a drug reaction, but numerous eosinophils (≥16/10 HPFs) are indicative of a drug reaction.75 Therefore, the histologic presentation
The histology of chronic GVHD is typically lichenoid in appearance and has significant overlap with idiopathic lichen planus (Fig. 7.120). These features are hyperkeratosis, hypergranulosis, irregular acanthosis, basal cell hydropic degeneration, cytoid body formation, pigmentary incontinence, and a variable bandlike lymphohistiocytic infiltrate that may obscure the dermal–epidermal interface. In contrast to idiopathic lichen planus, satellite cell necrosis is often present in the early phase of chronic GVHD, and the infiltrate sometimes contains plasma cells and eosinophils and may be less dense. Squamous metaplasia of the eccrine sweat ducts has been described.71,77
278 Lichenoid and interface dermatitis
Grade Feature
I Focal or diffuse vacuolar alteration of basal cells
II Vacuolar alteration of basal cells; spongiosis and dyskeratosis of epidermal cells
III Formation of subepidermal cleft in association with dyskeratosis and spongiosis
IV Complete loss of epidermis
Reproduced with permission from Lerner et al. (1974) Transplantation Proceedings, 6, 367–371.
The early changes of chronic GVHD may be indistinguishable from lichen planus. However, the dermal infiltrate is usually less conspicuous than that in lichen planus and sometimes contains plasma cells and eosinophils. The presence of satellite cell necrosis may be a diagnostic pointer toward chronic GVHD.
The late stage of chronic GVHD is characterized by epidermal atrophy with abolition of the ridge pattern and scarring of the superficial and deep dermis, with loss of the adnexal structures (Fig. 7.121) imparting a sclerodermoid feature including eosinophilic fasciitis, panniculitis, morphea-like changes, and lichen sclerosus.23,71,81,82 Features of the early stage of chronic GVHD, i.e., hydropic basal cell degeneration, cytoid body formation, and a chronic inflammatory cell infiltrate, may or may not be evident. Dermal mucin deposition has also been documented.83 Recently, GVHD associated angiomatosis has been associated with sclerodermoid chronic GVHD.84
Hepatic changes include bile duct atypia with necrosis, periportal inflammation, focal hepatocyte necrosis, and cholestasis.9 Gastrointestinal lesions show individual crypt cell necrosis accompanied by a mild chronic inflammatory cell infiltrate.71,85,86
In the absence of clinical information, it is usually not possible to distinguish the features of late chronic GVHD from morphea or systemic sclerosis.
The histologic features of the eruption of lymphocyte recovery are indistinguishable from acute GVHD. The differential diagnosis of acute GVHD includes engraftment syndrome. This syndrome can occur 10–14 days after transplantation but before peripheral lymphocytes are seen. It presents with a fever, hepatitis, intestinal symptoms, and an erythematous maculopapular eruption similar to acute GVHD. Some also require the presence of weight gain and pulmonary edema. Whether or not this represents a hypoacute GVHD is uncertain. The etiology is unknown, although it is postulated that the damage may be caused by cytokines released from recovering and degranulating neutrophils. G-CSF, GM-CSF, female sex, breast cancer, and other hematopoietic drugs have been implicated as risk factors for developing this condition.17,89
Differential diagnosis The features of acute GVHD can be reproduced by cytotoxic drugs such as cyclophosphamide and by radiotherapy. Viral infections also enter the differential diagnosis, as does an adverse drug reaction, for example, to antibiotic therapy. Although the presence of conspicuous eosinophils argues to some extent in favor of an adverse drug reaction, it may not be possible to distinguish drug reactions from acute GVHD histologically.75,87 In short, the regular practice of skin biopsy to differentiate between GVHD, drug reactions, chemotherapy effect, and viral infection may be questionable in some cases.
Acute GVHD may be indistinguishable from erythema multiforme and, in more severely affected patients, toxic epidermal necrolysis. Bile pigment deposition may be seen in the stratum corneum of a small percentage of patients with GVHD but not in cases of erythema multiforme.88
In summary, no histologic feature is pathognomonic for GVHD, and clinical correlation is essential. Therefore, in the appropriate clinical population, a positive biopsy can be very predictive despite subtle non-specific histologic findings. A negative biopsy is less reassuring. Some have advocated the use of a four-tier diagnostic system of no GVHD, possible GVHD, consistent with GVHD, and definite GVHD, a practical proposal that reflects the realities of daily practice.71
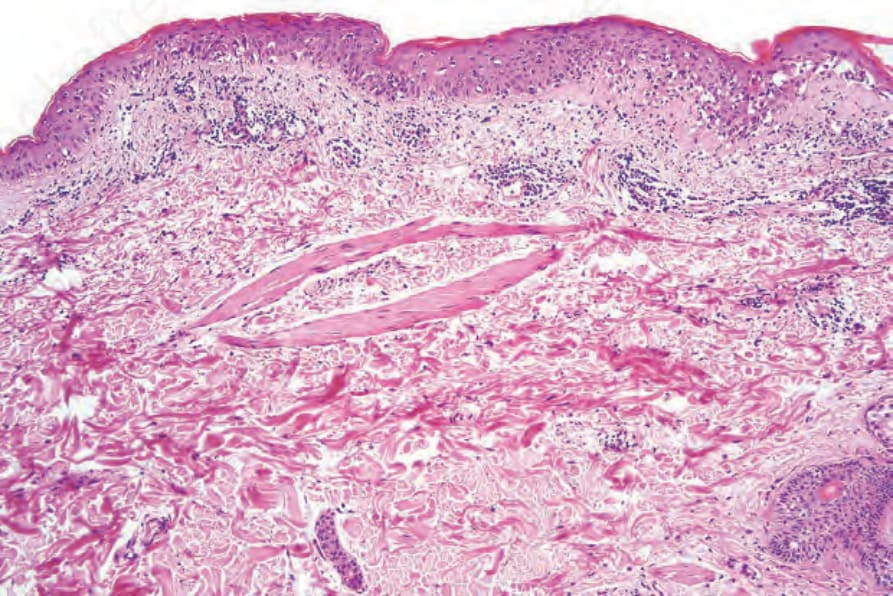
Fig. 7.116 Acute graft-versus-host disease: evolving lesion showing basal cell hydropic degeneration and scattered apoptotic keratinocytes. The dermis contains dilated blood vessels and a light perivascular chronic inflammatory cell infiltrate.
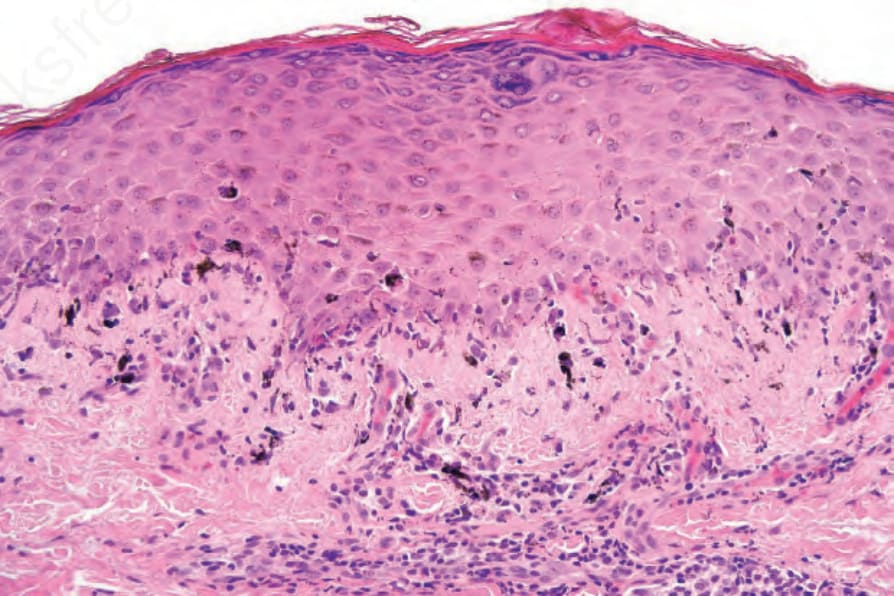
Fig. 7.120 Early chronic graft-versus-host disease: the hyperkeratosis, hypergranulosis, irregular acanthosis, and basal cell hydropic degeneration are indistinguishable from idiopathic lichen planus.
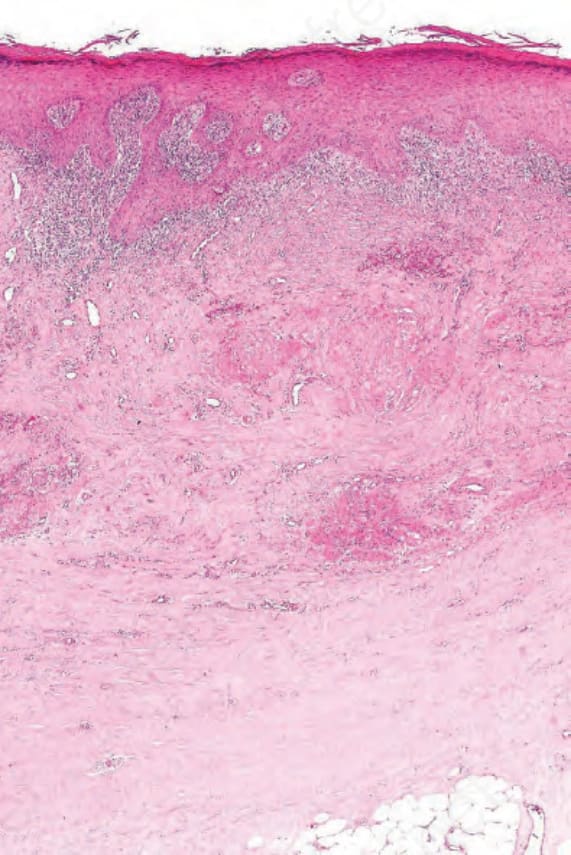
Fig. 7.121 Late chronic graft-versus-host disease: there is dense fibrosis of the dermis with tethering of the subcutaneous fat. Appendages are absent. These appearances are reminiscent of scleroderma.

Table 7.1 Grading of acute graft-versus-host disease