疾病定義與分類
- 先天性角化不良 (dyskeratosis congenita) 為一種罕見但重要的全身性疾病,預後差、死亡率高。
- 遺傳模式以 X-linked recessive 為主,主要侵犯男性(6:1);亦有 autosomal dominant 與 autosomal recessive 變異型。
- 核心特徵為一複雜三聯徵 (triad):皮膚色素沉著 (skin pigmentation)、指甲 (nail) 與黏膜 (mucosa) 異常。
- 惡性腫瘤發生率增加,包含血液系統與實質固態腫瘤。
臨床特徵
- 皮膚出現廣泛網狀色素沉著 (reticular pigmentation) 併 poikiloderma,初期最明顯於臉、頸與上胸「V」字領區,後續泛發。
- 兒童期指甲變為 dystrophic 且常脫落;可有掌蹠角化過度 (palmoplantar hyperkeratosis) 併多汗 (hyperhidrosis)、epiphora、早期落齒、齲齒、生長不良、稀疏毛髮、水皰性疹、淚管狹窄與智能不足。
- 肺纖維化 (pulmonary fibrosis) 導致瀰散能力下降。
- 重要併發症為侵犯口腔與肛門的癌前 leukoplakia,有顯著鱗狀細胞癌 (squamous cell carcinoma) 風險;尿道與陰道亦可能受侵犯。
- 血液學表現包含 thrombocytopenia、aplastic anemia、pancytopenia、myelodysplasia 與 acute myeloid leukemia。
- X-linked 變異型男性病情最嚴重;autosomal 變異型差異大,部分症狀輕微者可正常壽命。
- 預後不良主因與 aplastic anemia 併感染、惡性腫瘤與肺部併發症有關。
致病機轉 (Pathogenesis)
- 由與端粒 (telomere) 功能相關基因突變所致,受影響基因依遺傳模式而異。
- X-linked recessive 型源自 DKC1 基因突變(定位於 Xq28),多為 missense,導致 dyskerin 蛋白單一胺基酸取代;dyskerin 為核仁蛋白,負責 ribosomal RNA 之 pseudouridylation。
- 亦與 ACD、CTC1、DKC1、NHP2、NOP10、PARN、RTEL1、TERC、TERT、TINF2、WRAP53 等端粒功能相關基因相關;並非所有病例有已知遺傳原因。
- autosomal dominant 型與 telomerase RNA 成分 TERC、telomerase 酵素 TERT 突變相關(屬 shelterin 端粒保護複合體 TIN2)。
- autosomal recessive 型與調控 telomerase 之 NOP10/NHP2 基因突變相關。
- 顯著染色體不穩定,傾向發生重排;本病似源自 telomerase 活性缺陷,導致幹細胞更新或增殖活性受損;端粒明顯縮短且早年即出現。
組織病理特徵 (Histopathology)
- 色素變化的組織學表現為非特異性,僅見 pigmentary incontinence。
- 黏膜病灶切片顯示 acanthotic epithelium,可伴或不伴 dysplastic 變化;後者須特別小心排除 squamous cell carcinoma。
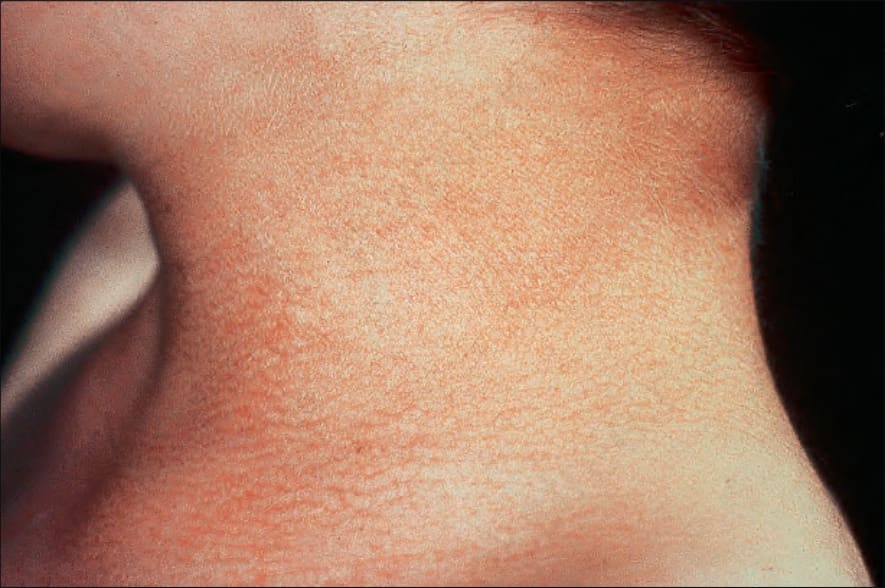
圖 7-104:Dyskeratosis congenita 頸部典型 poikiloderma 樣色素沉著。
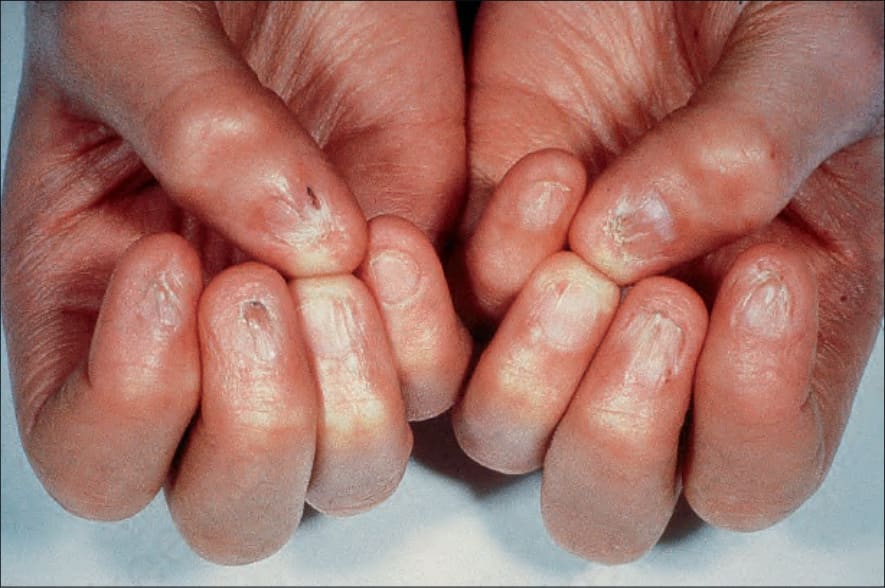
圖 7-105:Dyskeratosis congenita 指甲 dystrophy 併周圍皮膚明顯萎縮。