先天性角化不良 (Dyskeratosis Congenita)
臨床特徵 (Clinical Features)
-
先天性角化不良 (dyskeratosis congenita) 是一種罕見但重要的全身性疾病,預後不良且死亡率高。其遺傳模式主要為 X 染色體隱性遺傳 (X-linked recessive),多發生於男性 (男女比 6 : 1),不過也有體染色體顯性 (autosomal dominant) 與體染色體隱性 (autosomal recessive) 變異型被認可。
-
本病主要由皮膚色素沉著、指甲及黏膜異常所構成的一組複雜三聯徵 (triad) 組成。此外,惡性腫瘤的發生率亦增加,包括血液系統及實質固態腫瘤 (hematological and solid tumors)。
-
皮膚會出現廣泛的網狀色素沉著 (reticular pigmentation),並伴有異色症 (poikiloderma),起初最明顯地出現於臉部、頸部及上胸部的「V」字領區,但之後會變得全身泛發 (Fig. 7.104)。在兒童期,指甲會變得失養 (dystrophic),且常脫落 (Fig. 7.105)。也可能出現掌蹠角化過度 (palmoplantar hyperkeratosis) 並伴有多汗症 (hyperhidrosis)、溢淚 (epiphora)、牙齒早期脫落、齲齒 (caries)、生長不良、毛髮稀疏、大疱性疹 (bullous eruptions)、淚管狹窄 (lacrimal duct stenosis) 以及智能低下 (mental subnormality)。肺纖維化 (pulmonary fibrosis) 會造成擴散能力 (diffusion capacity) 下降。
-
特別侵犯口腔與肛門的癌前白斑症 (premalignant leukoplakia) 是一項重要的併發症,這些病灶有顯著的發展為鱗狀細胞癌 (squamous cell carcinoma) 的風險。尿道與陰道亦可能受侵犯。
致病機轉/分子 (Pathogenesis / Molecular)
-
涉及與端粒功能 (telomere function) 相關的 TINF2 及 WRAP53 基因。並非所有病例都有已知的遺傳病因。本病有顯著的染色體不穩定性 (chromosomal instability),並有明顯傾向發生染色體重排 (rearrangements)。因此,先天性角化不良似乎源自端粒酶活性 (telomerase activity) 缺陷,進而導致幹細胞 (stem cell) 更替或增殖活性受損。此論點獲得以下發現的支持:端粒明顯縮短,且此現象在很早的年齡即已發生。
-
體染色體顯性變異型同樣已被證實與端粒酶 RNA 組分 TERC、端粒酶酵素 TERT 的突變相關——兩者皆屬於 shelterin 端粒保護複合體 TIN2 的一部分。
-
最後,體染色體隱性變異型則與調控端粒酶的 NOP10/NHP2 基因突變相關。
組織病理特徵 (Histopathology)
-
色素變化的組織學特徵並無特異性,僅顯示色素失禁 (pigmentary incontinence)。
-
黏膜病灶的切片顯示棘層肥厚 (acanthotic) 的上皮,可伴有或不伴有異生性變化 (dysplastic changes)。在後者的情況下,必須極為謹慎以排除鱗狀細胞癌 (squamous cell carcinoma) 的存在。
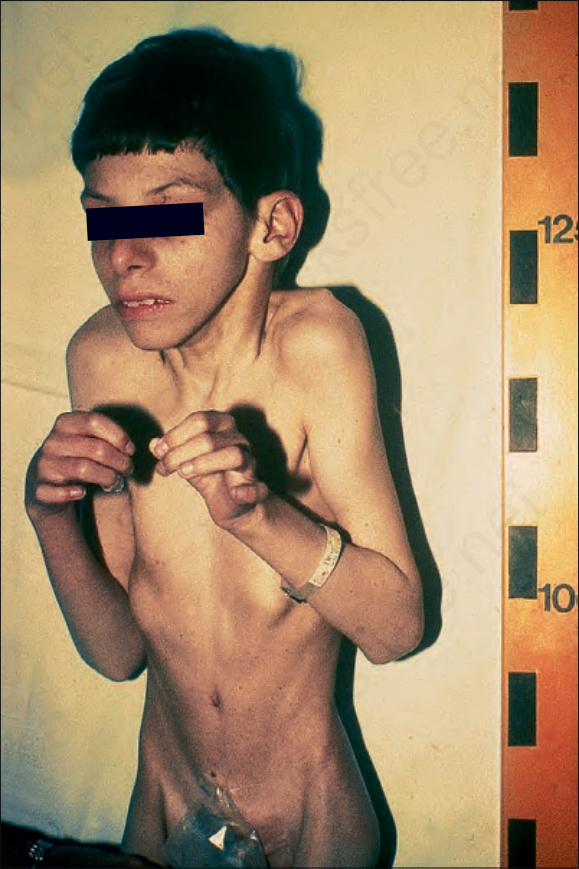
圖 7-103:Cockayne syndrome:其特徵包括突出的耳朵、下頜前突 (prognathism)、「鳥喙狀」鼻 (‘beaked’ nose) 及屈曲攣縮 (flexion contractures)。承蒙倫敦 Great Ormond Street 兒童醫院暨皮膚科學研究所 D. Atherton 醫師惠予提供。
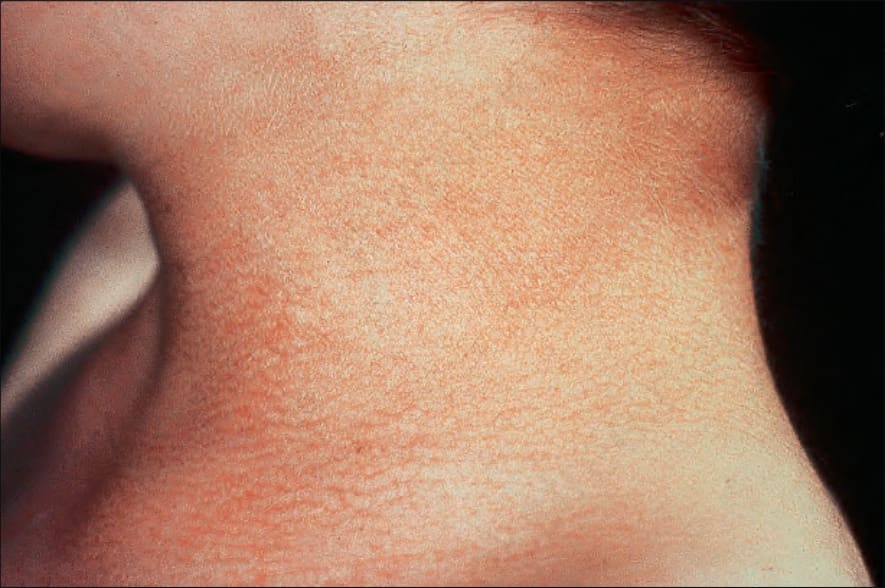
圖 7-104:先天性角化不良 (dyskeratosis congenita):頸部典型的異色症性色素沉著 (poikilodermatous pigmentation)。承蒙倫敦 Great Ormond Street 兒童醫院暨皮膚科學研究所 D. Atherton 醫師惠予提供。
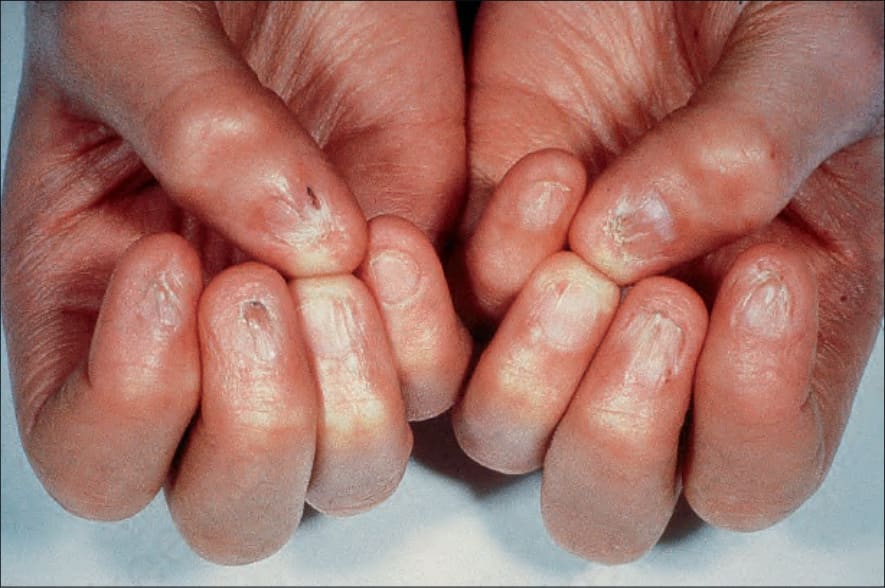
圖 7-105:先天性角化不良 (dyskeratosis congenita):可見指甲失養 (dystrophy),並伴有周圍皮膚的顯著萎縮 (atrophy)。承蒙倫敦 Great Ormond Street 兒童醫院暨皮膚科學研究所 D. Atherton 醫師惠予提供。