疾病定義與臨床特徵
- Rothmund-Thomson syndrome 為罕見、autosomal recessive 遺傳,見於亞洲人、黑人與白人;近期文獻提示男性偏好 (2 : 1)。
- 通常於出生後第 3–6 個月發病 (故又稱 ‘poikiloderma congenitale’),臉部呈網狀紅斑疹 (有時稱 marmoreal,大理石樣),後擴及四肢與臀部;軀幹與屈側通常倖免。
- 嬰兒有光敏感,皮膚病灶前常有日曬史;後續被網狀、線狀或點狀萎縮取代,伴 telangiectasia 與不等色素 (色素減退與沉著),異色變化最常見於日曬部位。
其他系統表現
- 脫髮 (尤其頭皮、眉、睫毛,女性多見),達 80% 病人。
- 嬰兒期可有慢性嘔吐與腹瀉;常見幼年型囊下白內障 (單或雙側)。
- 骨骼異常:矮小、骨質減少、病理性骨折、脫位、不規則幹骺端、異常骨小樑、髕骨點狀骨化;常見小手與短指。
- 特徵性 frontal bossing、saddle nose、prognathism;10–20% 病人 radii 缺如或畸形,可有 bifid 或缺如拇指。
- 甲營養不良、牙齒異常 (尤其錐形齒伴齲齒)、hypogonadism;伸側 (尤其手足) 可發展角化性疣狀病灶。智能不足通常非本病特徵。
致病機轉 (Pathogenesis)
- 部分病人與 RECQL4 基因 (DNA helicase family 成員) 突變相關;細胞遺傳分析顯示含 trisomy 8 之亞族鑲嵌現象。
- 確切潛在缺陷不明;多數研究未顯示對 UVA/UVB 異常敏感,惟偶有 UVB、UVC 照射後培養 fibroblasts 之 unscheduled DNA synthesis 減少報告。
- 較新研究提示 RECQL4 參與修復 UV 誘導之 DNA,並涉及 DNA 複製與骨骼生成。
診斷
- 無公認診斷試驗,診斷主要依異色症疹;RECQL4 突變篩檢可行並與部分臨床面向相關,但 USB1 等其他基因亦可能參與。
組織病理特徵 (Histopathology)
- 異色症組織學:hyperkeratosis、表皮萎縮、基底表皮細胞 liquefactive degeneration 與 telangiectasia。
- 可有色素失禁,表淺真皮有時見血管周圍慢性發炎浸潤;後者有時亦呈彈性組織碎裂與皮膚附屬器減少或缺如。鱗狀細胞癌呈典型特徵。
預後與惡性風險
- 與皮膚 squamous cell carcinoma、較罕見 basal cell carcinoma 之發生相關,亦有多灶性 Bowen disease 報告。
- 內部惡性腫瘤風險增加,尤其 tibial osteosarcoma 與多中心性 osteosarcoma (7–32%);曾有一病人合併十二指腸狹窄與環狀胰臟。
- 多數病人壽命大致正常。
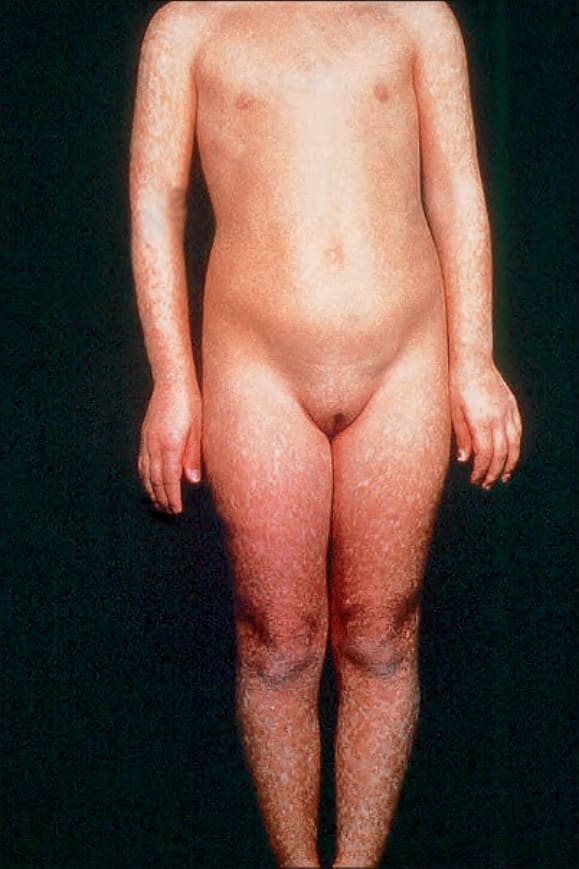
圖 7-99:Rothmund-Thomson syndrome,明顯斑駁色素沉著,主要侵犯周邊部位。
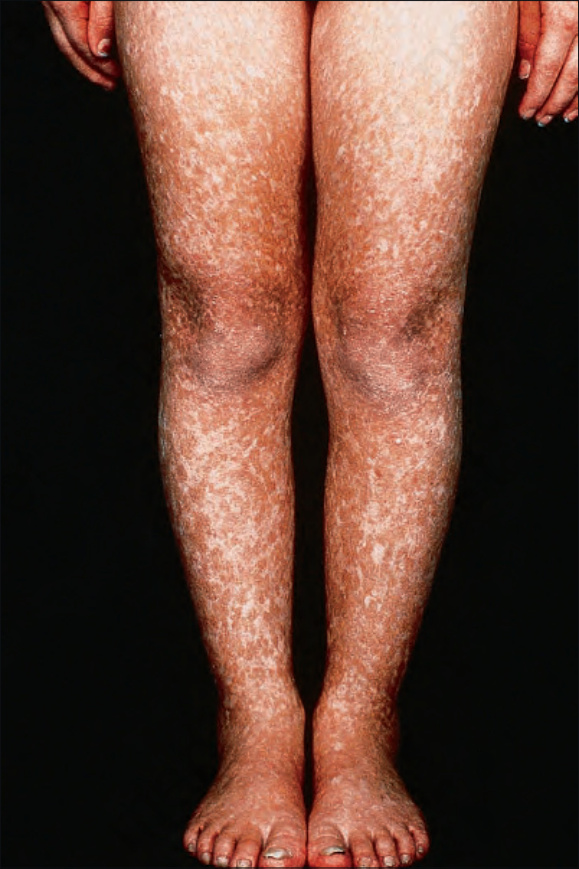
圖 7-100:Rothmund-Thomson syndrome,雙腿對稱受侵犯。
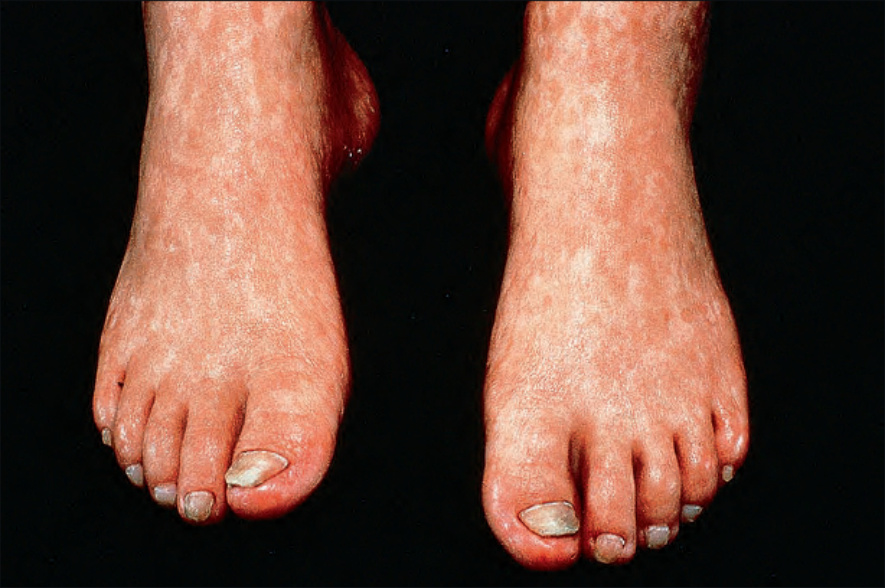
圖 7-101:Rothmund-Thomson syndrome,除色素沉著外尚有萎縮。