Rothmund-Thomson syndrome
Rothmund-Thomson syndrome
Clinical features This rare syndrome, which has been described in Asians and blacks as well as Caucasians, has an autosomal recessive mode of inheritance. In contrast to the earlier finding of an equal sex incidence, the more recent literature suggests a predilection for males (2 : 1).1,2 It usually presents between the third and sixth months of life (hence the term ‘poikiloderma congenitale’) as a reticulated, erythematous rash – sometimes described as marmoreal (L. marmor, marble) – on the face, which eventually spreads to involve the extremities and the buttocks (Figs 7.99–7.101).2 The trunk and flexural aspects are usually spared.1 Affected infants are photosensitive and, therefore, there is often a history of sun exposure before the development of skin lesions.3,4 This is later replaced by reticular, linear, or punctate foci of atrophy.4 Telangiectasia is present and areas of hypo- and hyperpigmentation may be noted. The poikilodermatous change is seen most frequently at sun-exposed sites.1
A variety of other manifestations may be observed, including variable alopecia particularly involving the scalp, eyebrows, and eyelashes, and seen most often in females. This is present in up to 80% of patients.1 Gastrointestinal problems including chronic emesis and diarrhea may be seen in infancy.2 Juvenile, subcapsular (unilateral or bilateral) cataracts are common and skeletal abnormalities include short stature, osteopenia, pathological fractures, dislocations, irregular metaphyses, abnormal trabeculation, and stippled ossification of the patellae.2 Small hands with shortened digits are frequently seen.5 Frontal bossing, saddle nose, and prognathism are characteristic.1 Absent or malformed radii are seen in 10–20% of patients and bifid or absent thumb may also be present.1,2,6 Nail dystrophy, dental abnormalities (particularly conical-shaped teeth with caries), and hypogonadism may also be detected. Hyperkeratotic warty or verrucous lesions sometimes develop on the extensor surfaces, particularly overlying joints and especially the feet and hands.6 While occasionally reported, mental retardation is not usually a feature of this syndrome.1 The disease is associated with the development of cutaneous squamous cell carcinoma and more rarely basal cell carcinoma.2,4,7,8 Multifocal Bowen disease has also been described.8
There is also an increased risk of internal malignancies, particularly tibial osteosarcoma and multicentric osteosarcoma (7–32%).1,2,7,9–12 An association with duodenal stenosis and annular pancreas has been described in one patient.13 The life span of most patients, however, is generally normal.
271 Interface dermatoses
Pathogenesis and histologic features Rothmund-Thomson syndrome, in some patients at least, has been shown to be associated with a mutation in the RECQL4 gene, a member of the DNA helicase family (see Bloom syndrome).10,14–17 Cytogenetic analysis has revealed mosaicism in a subpopulation including trisomy 8.2 The underlying defect in Rothmund-Thomson syndrome is unknown. While most investigations have failed to demonstrate abnormal sensitivity to UVA or UVB, there have been occasional recent reports of reduced unscheduled DNA synthesis following irradiation of cultured fibroblasts with UVB and UVC.3,18 More recent studies suggest a role for RECQL4 in repairing DNA induced by UV irradiation.19 Other investigations have demonstrated that RECQL4 is also involved in DNA replication and skeletogenesis.20–24
No recognized diagnostic test for this disorder is available, and diagnosis is based primarily on the polioderma rash.17 Mutational screening for the RECQL4 gene is possible and can correlate with certain aspects of the syndrome, but additional genes such as USB1 may also be involved.25,26
high-pitched, squeaky (so-called ‘Mickey Mouse’) voice is sometimes a feature.6 Male infertility is common.3 Patients may suffer impaired concentration, short-term memory, and general mental organizational disability.5
The histologic features of poikiloderma include hyperkeratosis, epidermal atrophy, liquefactive degeneration of the basal epidermal cells, and telangiectasia. Pigmentary incontinence may be present and a perivascular chronic inflammatory cell infiltrate is sometimes evident in the superficial dermis. The latter sometimes also shows elastic tissue fragmentation and depletion or absence of cutaneous appendages.1 The squamous cell carcinomas show typical features.
An examination of scalp has revealed hypopigmented vellus hairs without cortices.6
Bloom syndrome is typified by an inherent propensity to chromosomal abnormalities, in particular, sister chromatid exchange. There is an associated increased incidence of most malignancies, especially acute leukemia, non-Hodgkin lymphoma, colon carcinoma, breast carcinoma, and cutaneous squamous cell carcinoma. Patients are prone to develop multiple primary tumors, which often develop at an early age (third decade). As a result, death by age 30 usually occurs due to cancer.4,5,7 They may also suffer immunodeficiency (diminished IgG, IgA, IgM) and are therefore at an increased risk of childhood infections, pulmonary infections, and chronic lung disease.6,8 There is also an elevated risk of adult onset-like diabetes mellitus.9
Pathogenesis and histologic features The gene for Bloom syndrome, BLM, which has been mapped to 15q21.3, is a member of the RecQ helicase protein family, responsible for unwinding DNA and RNA.5,10–16 It has been identified as representing part of the BRCA1-associated genome surveillance complex, which is mutated in families with hereditary breast cancer.17 The protein functions as a 3′–5′ DNA helicase and may be involved specifically in allowing sister chromatid separation during mitosis.13,18 DNA helicases have essential roles in genetic recombination, transcription, DNA replication, and repair.5,16 Mutation of the BLM gene results in genomic instability. BLM normally interacts with BRAFT and FANCOM complexes. The BRAFT complex is involved in helicase activity, whereas the FANCOM complex is crucial for sister chromatid exchange.5 Bloom syndrome is associated with increased sensitivity to alkylating agents, increased spontaneous chromosome breakages, increased interchromatid exchange (including sister chromatid exchange, 6–10-fold), increased somatic cell mutation frequency, and reduced replication fork elongation rate.4,5,12 Mutations include missense, nonsense, frameshift, and genomic deletions, most of which result in premature translation
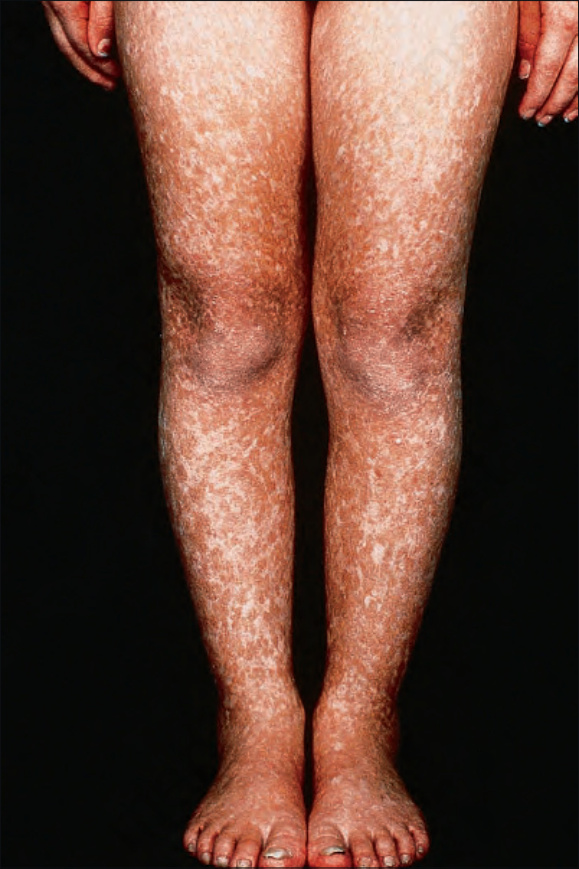
Fig. 7.100 Rothmund-Thomson syndrome: there is symmetrical involvement of the legs. By courtesy of the Institute of Dermatology, London, UK.
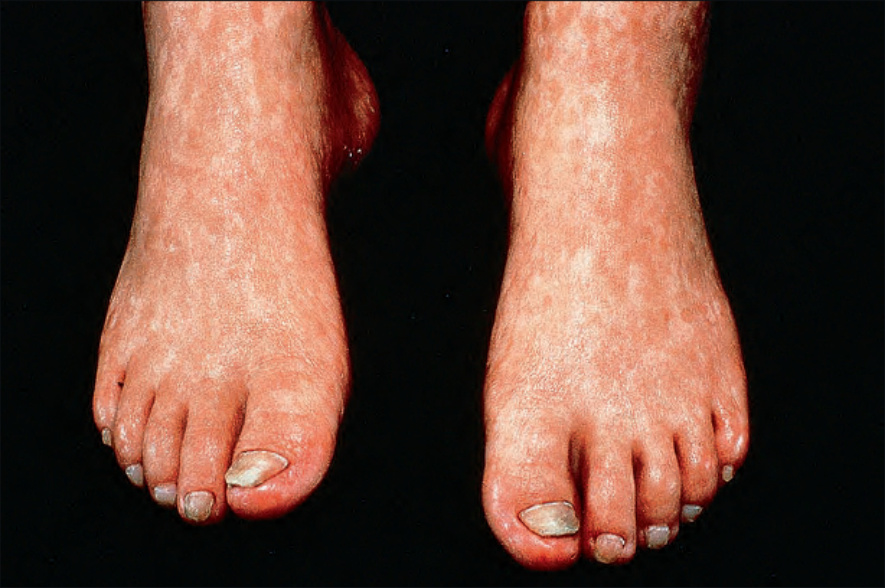
Fig. 7.101 Rothmund-Thomson syndrome: there is atrophy in addition to hyperpigmentation. By courtesy of the Institute of Dermatology, London, UK.
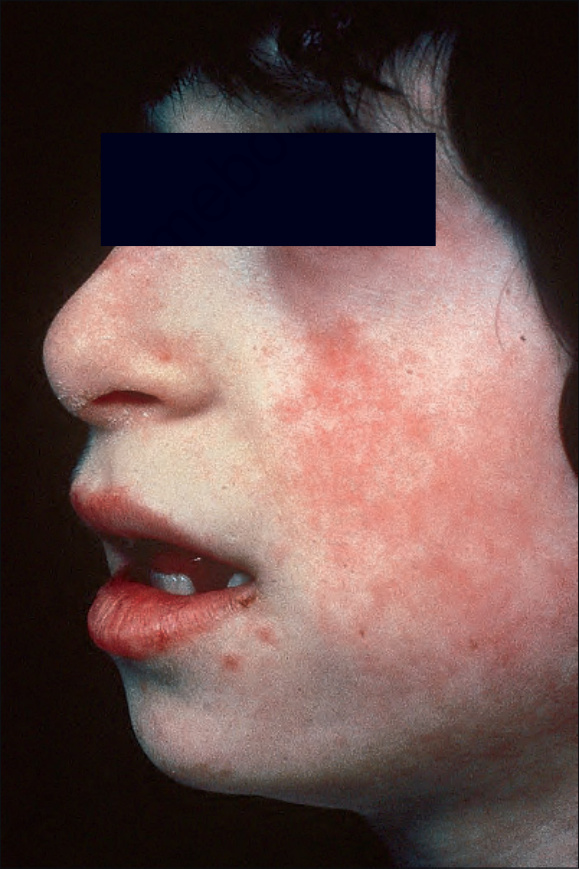
Fig. 7.102 Bloom syndrome: characteristic facies includes ‘pinched’ features. Marbled erythema of the cheek and crusted lesions involving the lower lip. By courtesy of D. Atherton, MD, Institute of Dermatology and Children’s Hospital at Great Ormond Street, London, UK.
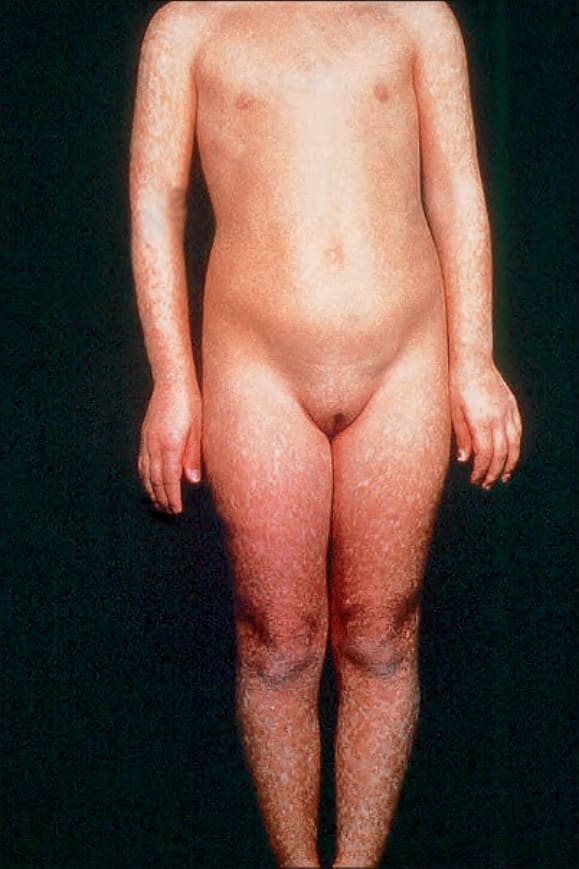
Fig. 7.99 Rothmund-Thomson syndrome: there is a marked mottled hyperpigmentation predominantly affecting the peripheries. By courtesy of the Institute of Dermatology, London, UK.