Rothmund-Thomson 症候群 (Rothmund-Thomson syndrome)
臨床特徵 (Clinical Features)
-
此罕見症候群除曾見於高加索人外,亦曾於亞洲人與黑人中被描述,遺傳模式為體染色體隱性 (autosomal recessive)。相較於早期所發現的兩性發生率相等,較近期的文獻顯示其有偏好男性的傾向 (2:1)。
-
通常於出生後第三至第六個月之間表現(因此有「先天性異色症 (poikiloderma congenitale)」一詞),表現為臉部的網狀、紅斑性皮疹——有時被描述為大理石樣 (marmoreal;L. marmor,大理石)——最終擴散至侵犯四肢與臀部 (Figs 7.99–7.101)。軀幹與屈側通常不受侵犯。
-
受影響的嬰兒具有光敏感性 (photosensitive),因此在皮膚病灶發生之前常有日曬史。其後此皮疹被網狀、線狀或點狀的萎縮病灶所取代。可見毛細血管擴張 (telangiectasia),亦可注意到色素減退與色素增加的區域。異色症 (poikilodermatous) 變化最常見於日曬部位。
-
可觀察到多種其他表現,包括程度不一的禿髮 (alopecia),尤其侵犯頭皮、眉毛與睫毛,且最常見於女性。此見於高達 80% 的患者。嬰兒期可見胃腸道問題,包括慢性嘔吐與腹瀉。幼年型、囊膜下 (subcapsular)(單側或雙側)白內障 (cataracts) 常見,骨骼異常則包括身材矮小、骨質減少 (osteopenia)、病理性骨折、脫位、不規則幹骺端 (metaphyses)、異常骨小樑 (trabeculation),以及髕骨的點狀骨化 (stippled ossification)。常見手部小且手指短縮。額部隆凸 (frontal bossing)、鞍鼻 (saddle nose) 與下頜前突 (prognathism) 為其特徵。10–20% 的患者可見橈骨 (radii) 缺如或畸形,亦可能出現拇指分叉或缺如。亦可偵測到指甲萎縮 (nail dystrophy)、牙齒異常(尤其是伴齲齒的圓錐狀牙齒),以及性腺功能低下 (hypogonadism)。有時於伸側表面發展出過度角化的疣狀 (warty 或 verrucous) 病灶,尤其覆蓋於關節上方,特別是足與手。雖偶有報告,智能障礙通常並非此症候群的特徵。本病與皮膚鱗狀細胞癌 (squamous cell carcinoma) 之發生有關,較罕見地與基底細胞癌 (basal cell carcinoma) 有關。亦曾有多病灶 Bowen 氏病 (multifocal Bowen disease) 的描述。
-
亦有內臟惡性腫瘤風險增加,尤其是脛骨骨肉瘤 (tibial osteosarcoma) 與多中心性骨肉瘤 (multicentric osteosarcoma)(7–32%)。曾於一名患者中描述與十二指腸狹窄 (duodenal stenosis) 及環狀胰腺 (annular pancreas) 之關聯。然而,大多數患者的壽命一般正常。
271 Interface dermatoses
致病機轉與組織學特徵 (Pathogenesis and Histologic Features)
-
Rothmund-Thomson 症候群,至少在部分患者中,已被證明與 RECQL4 基因(DNA helicase 家族的一員,參見 Bloom syndrome)的突變有關。細胞遺傳學分析已揭示一個亞群中的鑲嵌現象 (mosaicism),包括第 8 對染色體三體 (trisomy 8)。Rothmund-Thomson 症候群的潛在缺陷尚未明瞭。雖然大多數研究未能證明對 UVA 或 UVB 有異常敏感性,但近期偶有報告指出,培養的纖維母細胞 (fibroblasts) 經 UVB 與 UVC 照射後非排程 DNA 合成 (unscheduled DNA synthesis) 減少。較近期的研究顯示 RECQL4 在修復 UV 照射所誘發之 DNA 中扮演角色。其他研究已證明 RECQL4 亦參與 DNA 複製與骨骼發生 (skeletogenesis)。
-
此疾患目前無公認的診斷檢測,診斷主要基於異色症 (polioderma) 皮疹。RECQL4 基因的突變篩檢是可行的,且可與此症候群的某些面向相關聯,但其他基因如 USB1 亦可能涉及其中。
-
高音調、尖細(所謂「米老鼠 (‘Mickey Mouse’)」)的嗓音有時為一項特徵。男性不孕 (infertility) 常見。患者可能罹患注意力受損、短期記憶障礙與一般心智組織能力障礙。
-
異色症的組織學特徵包括過度角化 (hyperkeratosis)、表皮萎縮 (epidermal atrophy)、基底表皮細胞的液化變性 (liquefactive degeneration),以及毛細血管擴張 (telangiectasia)。可能存在色素失禁 (pigmentary incontinence),且淺層真皮中有時可見血管周圍慢性發炎細胞浸潤。後者有時亦顯示彈性組織碎裂,以及皮膚附屬器 (cutaneous appendages) 的減少或缺如。鱗狀細胞癌則顯示典型特徵。
-
頭皮的檢查已揭示無皮質 (cortices) 的色素減退毳毛 (vellus hairs)。
-
Bloom syndrome 的典型特徵為染色體異常的內在傾向,尤其是姊妹染色分體互換 (sister chromatid exchange)。其伴隨大多數惡性腫瘤的發生率增加,尤其是急性白血病 (acute leukemia)、非何杰金氏淋巴瘤 (non-Hodgkin lymphoma)、結腸癌 (colon carcinoma)、乳癌 (breast carcinoma) 與皮膚鱗狀細胞癌。患者易於發展出多發性原發腫瘤,且常於早年(第三個十年)發生。因此,30 歲前死亡通常因癌症而發生。患者亦可能罹患免疫缺陷(IgG、IgA、IgM 降低),因此罹患兒童感染、肺部感染與慢性肺病的風險增加。亦有成人型糖尿病 (adult onset-like diabetes mellitus) 風險升高。
-
致病機轉與組織學特徵 Bloom syndrome 的基因 BLM 已被定位至 15q21.3,為 RecQ helicase 蛋白家族的一員,負責解開 DNA 與 RNA。其已被確認為 BRCA1 相關基因體監視複合體 (BRCA1-associated genome surveillance complex) 的一部分,此複合體在遺傳性乳癌家族中發生突變。該蛋白以 3′–5′ DNA helicase 的方式運作,可能特別參與允許有絲分裂 (mitosis) 期間姊妹染色分體的分離。DNA helicase 在基因重組、轉錄、DNA 複製與修復中具有不可或缺的角色。BLM 基因突變導致基因體不穩定。BLM 正常時與 BRAFT 及 FANCOM 複合體交互作用。BRAFT 複合體參與 helicase 活性,而 FANCOM 複合體對姊妹染色分體互換至關重要。Bloom syndrome 與對烷化劑 (alkylating agents) 敏感性增加、自發性染色體斷裂增加、染色分體間互換增加(包括姊妹染色分體互換,6–10 倍)、體細胞突變頻率增加,以及複製叉延伸速率 (replication fork elongation rate) 降低有關。突變類型包括錯義 (missense)、無義 (nonsense)、移碼 (frameshift) 與基因體缺失,其中大多數導致過早轉譯 (premature translation)。
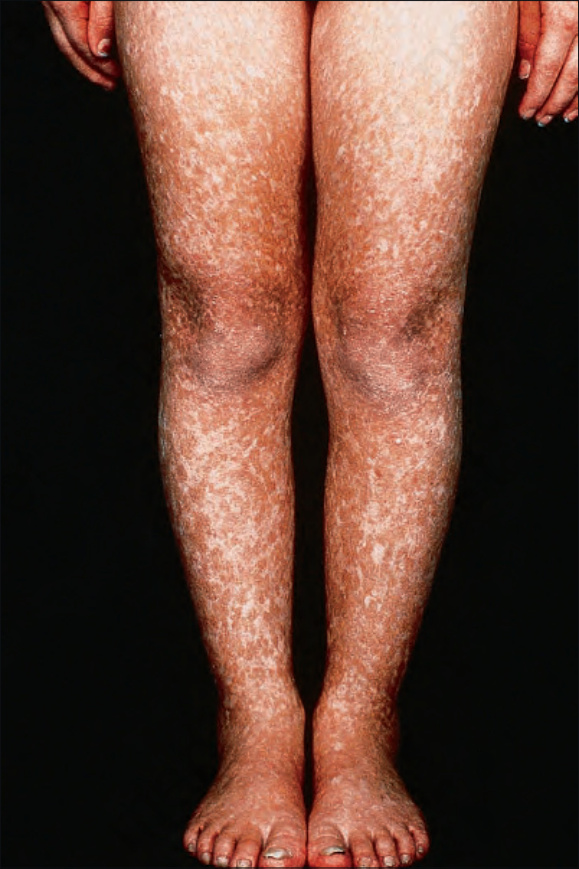
圖 7-100:Rothmund-Thomson 症候群:雙腿呈對稱性侵犯。承蒙英國倫敦皮膚科研究所 (Institute of Dermatology) 提供。
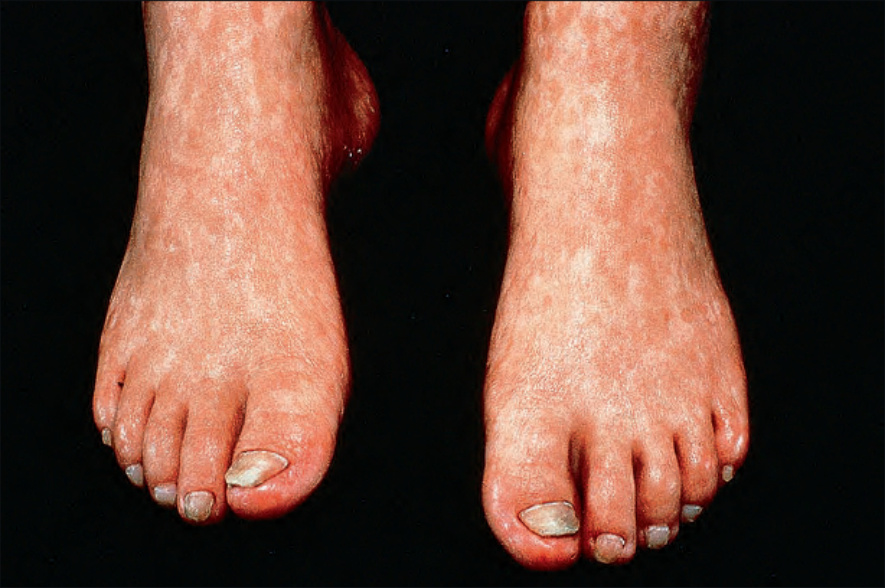
圖 7-101:Rothmund-Thomson 症候群:除色素增加外,尚有萎縮 (atrophy)。承蒙英國倫敦皮膚科研究所 (Institute of Dermatology) 提供。
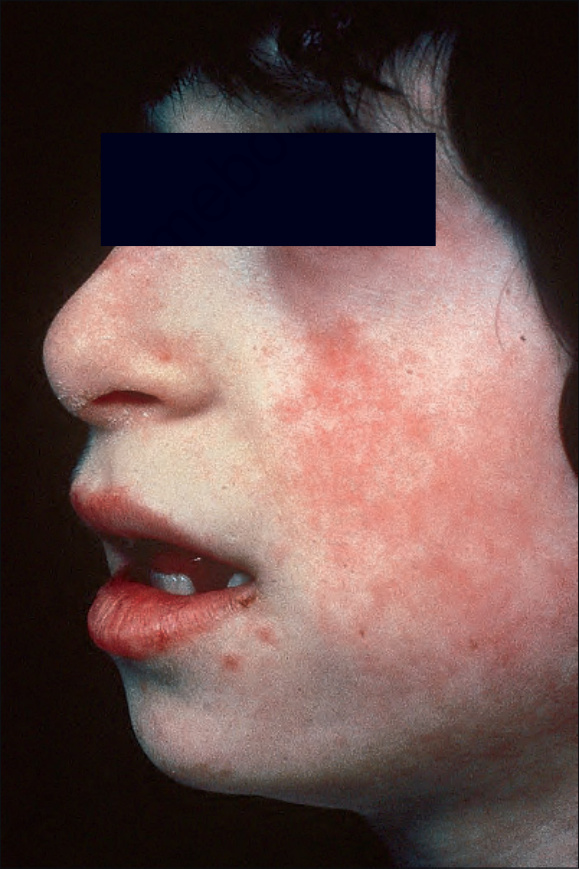
圖 7-102:Bloom syndrome:特徵性面容包括「捏縮 (‘pinched’)」的容貌。臉頰的大理石樣紅斑 (marbled erythema) 與侵犯下唇的結痂 (crusted) 病灶。承蒙英國倫敦皮膚科研究所暨大奧蒙德街兒童醫院 (Institute of Dermatology and Children’s Hospital at Great Ormond Street) D. Atherton 醫師 (MD) 提供。
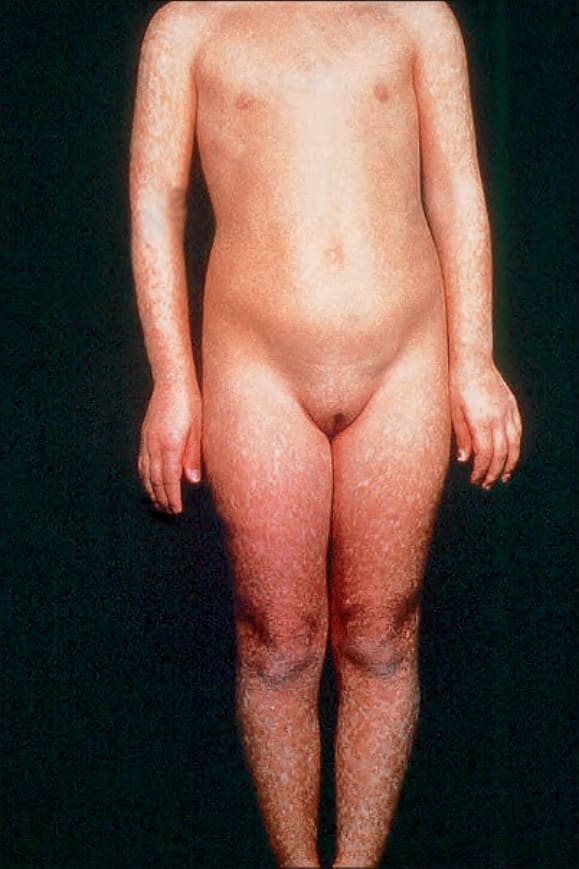
圖 7-99:Rothmund-Thomson 症候群:有顯著的斑駁色素增加 (mottled hyperpigmentation),主要影響周邊部位。承蒙英國倫敦皮膚科研究所 (Institute of Dermatology) 提供。