疾病定義與分類
- 先天性厚甲症 (pachyonychia congenita, PC) 為一群 autosomal dominant 遺傳性皮膚病。
- 與 keratin 6A、6B、6C、keratin 16 或 keratin 17 基因突變相關,影響多種外胚層結構:甲床、掌蹠皮膚、口腔黏膜、牙齒與毛皮脂腺單位。
- 歷史分型:PC type 1 (Jadassohn-Lewandowsky) 與 PC type 2 (Jackson-Lawler)。
- PC type 1:K6a/K16 突變,表現 PPK 與口腔白色角化症 (oral leukokeratosis)。
- PC type 2:K6b/K17 突變,表現毛皮脂腺單位相關病理(cysts)與新生兒牙齒。
- 後依受影響 keratin 重新分類:PC-6a、PC-6b、PC-6c、PC-16、PC-17。
臨床表現 (Clinical features)
- 三大主徵見於九成以上病人:趾甲失養 (toenail dystrophy)、局灶性角化症 (focal keratoderma)、蹠部疼痛 (plantar pain)。
- 甲下過度角化致甲板抬高、增厚、變暗與彎曲;趾甲及拇指、食指甲受累最嚴重;嬰兒期甲床紅斑可先於甲失養。
- 受壓部位形成厚黃色角化(胼胝)、裂隙與摩擦性水皰,尤以夏季為甚;蹠部劇痛影響行走。
- 掌蹠多汗 (palmoplantar hyperhidrosis) 與甲床感染。
- 其他:膝肘毛囊性過度角化、oral leukokeratosis、囊腫、出生即有的牙齒;舌與口腔黏膜斑塊狀白色增厚區。
- 喉部侵犯可致聲音嘶啞,嬰兒期甚至致命性呼吸道阻塞。
- keratin 17 突變病人發展 steatocystomas 及/或其他毛皮脂腺囊腫,並造成出生時牙齒萌發。
致病機轉 (Pathogenesis)
- 多數致病突變為 heterozygous missense 突變或小段 insertion/deletion,破壞細胞骨架致細胞脆弱。
- 突變 keratin 之不同表現模式對應病灶分布的差異。
- keratin 6a 突變(主要甲 keratin)另影響口腔黏膜。
- KRT16、KRT17 突變可分別見於單純局灶性 PPK 或 steatocystoma multiplex 而無其他 PC 表現;KRT6c 突變相關之局灶性角化症較其他型輕微。
組織病理特徵 (Histology)
- 掌蹠表皮:hyperkeratosis、acanthosis 與斑塊狀 hypergranulosis,含大而畸形的 keratohyalin granules,但無 epidermolysis。
- 毛囊病灶:毛囊口栓塞,鄰近 hyperkeratosis、parakeratosis 與 acanthosis,伴蒼白角質細胞。
- 皮膚與口腔黏膜的棘層上上皮呈特徵性蒼白胞質與嗜酸性內含物;超微結構對應突變 keratin filaments 核周凝聚與周邊胞質蒼白染色或空泡化。
- 囊腫可為角質性 epidermoid cysts、eruptive vellus hair cysts 或真正的 steatocystomas。
鑑別診斷 (Differential diagnosis)
- white sponge nevus(keratin 4、13 突變):同樣可見蒼白上皮伴嗜酸性內含物。
- twenty-nail dystrophy:frizzled 6 gene (FZD6) autosomal recessive 突變。
- 僅有 pachyonychia 樣甲變化而無其他 PC 症狀者:connexin 30 heterozygous missense 突變。
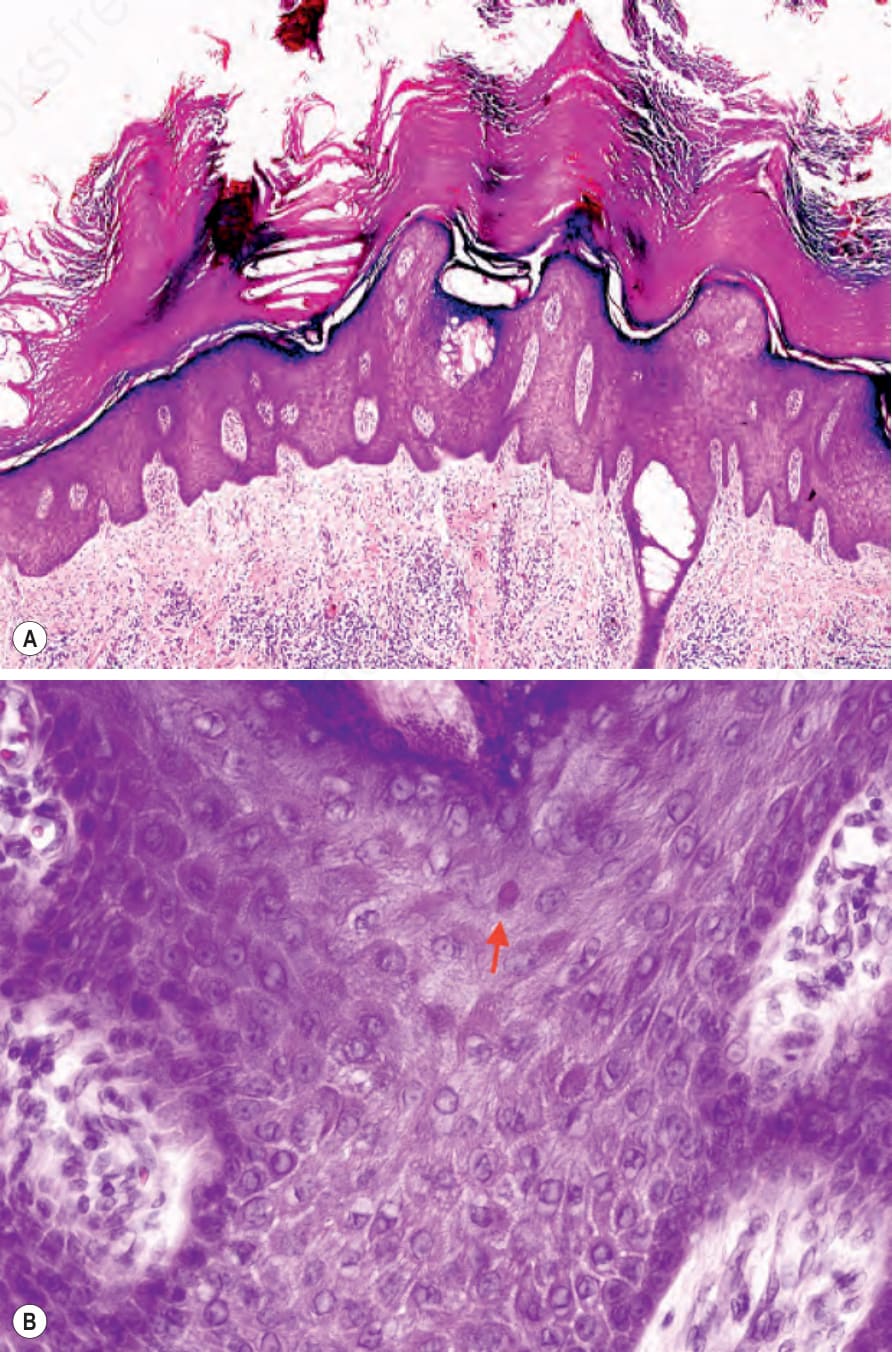
圖 3-120:Pachyonychia congenita:(A) 掌側皮膚顯示大量 hyperkeratosis、hypergranulosis 與 acanthosis;(B) 棘層上角質細胞具特徵性蒼白胞質與嗜酸性內含物。
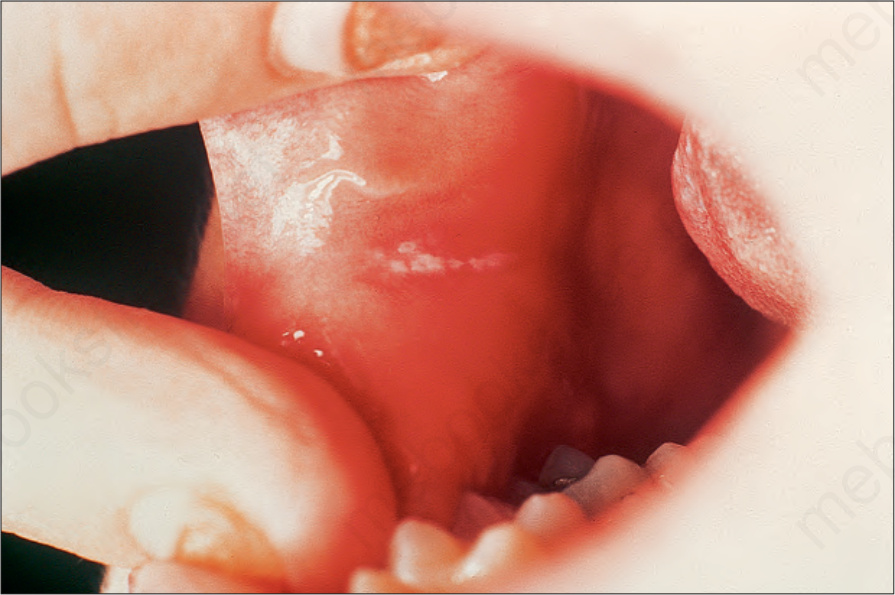
圖 3-119:Pachyonychia congenita:頰黏膜白斑 (leukoplakia) 為常見伴隨特徵。