Pachyonychia congenita
Pachyonychia congenita
Pachyonychia congenita (PC) comprises a group of autosomal dominant genodermatoses related to mutations in the genes for keratin 6A, 6B, 6C, keratin 16, or keratin 17 affecting a number of ectodermal structures, including nail bed, volar skin, oral mucosae, teeth, and pilosebaceous units.1–3
100 Disorders of keratinization
A
A
B
B
Fig. 3. 117 Pachyonychia congenita: (A) there is gross nail deformity with transverse arching of the distal portion. Although the nail plate appears to be thickened, most of the changes are, in fact, due to massive hyperkeratosis of the nail bed, resulting in elevation and bending of the nail plate; (B) in this view, the subungual hyperkeratosis is more obvious.
darkening, and bending of the nail plate (Fig. 3.117). All digits may or may not be involved and toenails and nails of the thumb and index fingers are most severely affected. Erythema of the nail bed can precede nail dystrophy in infancy. Thick yellow keratoses develop on sites of pressure forming calluses (Fig. 3.118), fissures, and frictional blisters, particularly during the summer. On the soles, severe pain, which makes walking difficult, is observed. Patients also suffer from palmoplantar hyperhidrosis and nail bed infections.
Historically, PC has been classified as PC type 1 (Jadassohn-Lewandowsky) and PC type 2 (Jackson-Lawler).4–6 As originally described, PC type 1 is associated with mutations in genes encoding the dimerizing pair of K6a/ K16 and presents with PPK and oral leukokeratosis, while PC type 2 is caused by mutations of K6b/K17 and should feature pathology associated with pilosebaceous units (cysts) and neonatal teeth.7 However, a large genotype/phenotype analysis resulted in a classification system based on the affected keratin, e.g., PC-6a, PC-6b, PC6c, PC-16, PC-17.8 Further information on the subject is given by the International PC Consortium (IPCC) (www.pachyonychia.org).
Clinical features Clinically, three cardinal symptoms are present in more than 90% of all patients with PC: toenail dystrophy, focal keratoderma, and plantar pain.3,8,9 Subungual hyperkeratosis of the nail leads to elevation, thickening,
Additional diagnostic findings include follicular hyperkeratoses on the knees and elbows, oral leukokeratosis, palmoplantar hyperhidrosis, cysts, and natal teeth.9 Patchy white thickened areas are seen on the tongue and oral mucosa (Fig. 3.119).10 Laryngeal involvement may produce hoarseness and in infancy even fatal respiratory obstruction.11 Patients with keratin 17 mutations develop steatocystomas and/or other forms of pilosebaceous cysts. Keratin 17 mutations are also responsible for erupted teeth present at birth.9
Pathogenesis and histologic features The majority of causative mutations in PC-related keratins are heterozygous missense mutations or small insertions/deletion mutations that disrupt the cytoskeleton resulting in cell fragility.12 The different expression patterns of the mutant keratins correlate with the variable distribution of lesions.13–16
101 Palmoplantar keratoderma
A
B
keratohyalin granules are present but epidermolysis is missing (Fig. 3.120).1,15 The follicular lesions show plugging of the ostia with adjacent hyperkeratosis, parakeratosis, and acanthosis with pale keratinocytes (Fig. 3.121).4 Suprabasal epithelial of the skin and oral mucosa show a characteristic pale cytoplasm and eosinophilic inclusions (Fig. 3.122). At ultrastructural level, these findings correlate with perinuclear condensation of mutated keratin filaments and pale staining or vacuolization of the peripheral cytoplasm.8,22 Cysts may be keratinous epidermoid cysts, eruptive vellus hair cysts or true steatocystomas.
Mutations in the prominent nail keratin, keratin 6a, additionally affect the oral mucosa.17 Keratin 17 is constitutively expressed in the pilosebaceous unit, but not to the same extent as in palmoplantar skin and mucosae.18,19 Mutations in KRT16 and KRT17 have been reported in patients with isolated focal PPK or steatocystoma multiplex, respectively, without any other PC manifestations.20 Mutations in KRT6c seem to be associated with a much milder focal keratoderma when compared with any other of the four PC types.21
Histology of the affected palmoplantar epidermis shows hyperkeratosis, acanthosis, and patchy hypergranulosis, in which large and malformed
Differential diagnosis The same histologic pattern of pale epithelia with eosinophilic inclusions can be also found in white sponge nevus where recurrent white plaques develop on oral and other mucosal sites as a consequence of keratin 4 and 13 mutations. Other differential diagnoses include twenty-nail dystrophy caused by autosomal recessive mutation of the frizzled 6 gene (FZD6).23 In patients with pachyonychia-like nail changes devoid of any other symptoms of PC, heterozygous missense mutations in the connexin 30 have been demonstrated.24
102 Disorders of keratinization
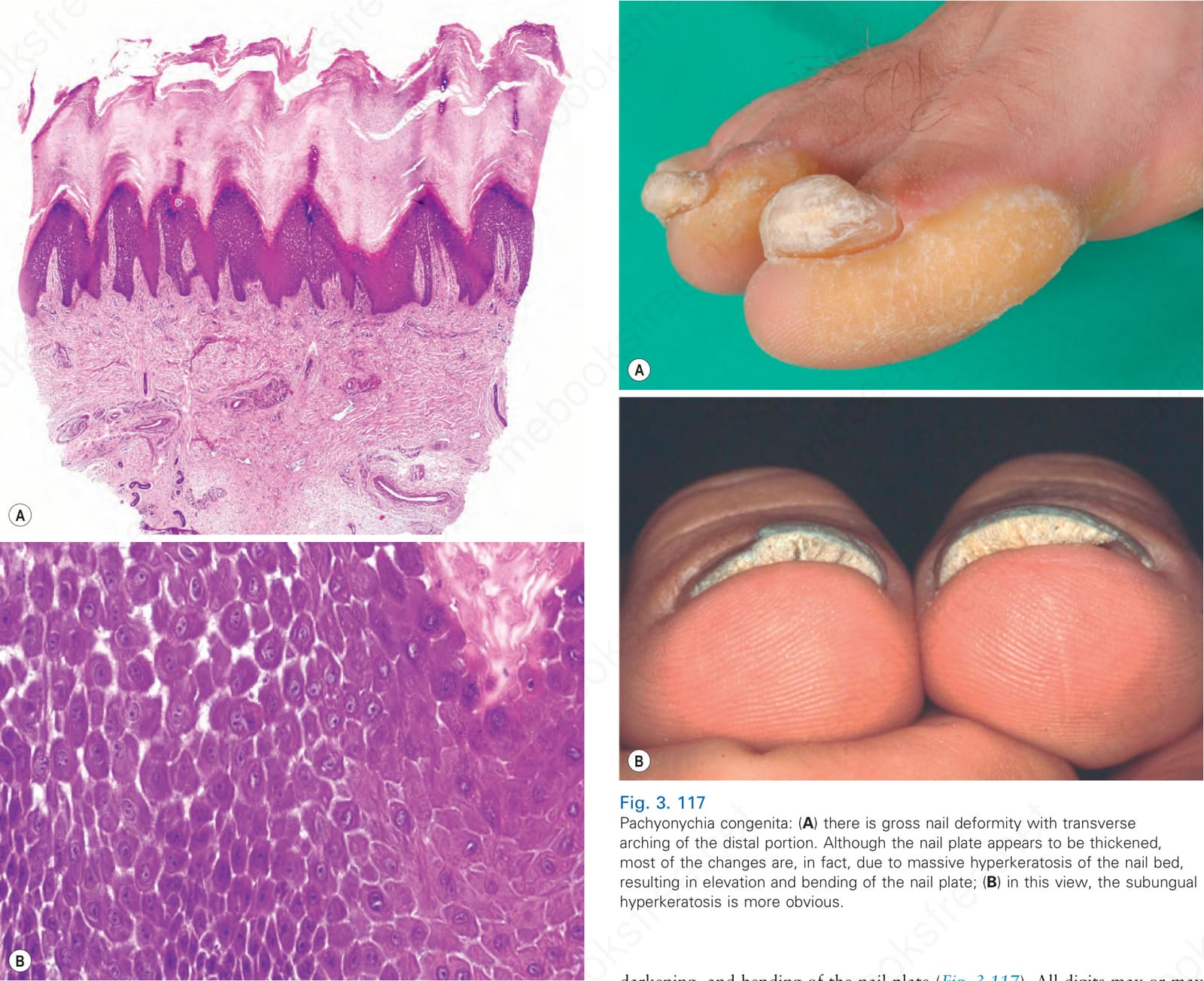
Fig. 3.116 Carvajal-Huerta syndrome: (A) massive orthohyperkeratosis, acanthosis, and papillomatosis; (B) partial dehiscence of suprabasal keratinocytes and characteristic widening of the intercellular spaces.
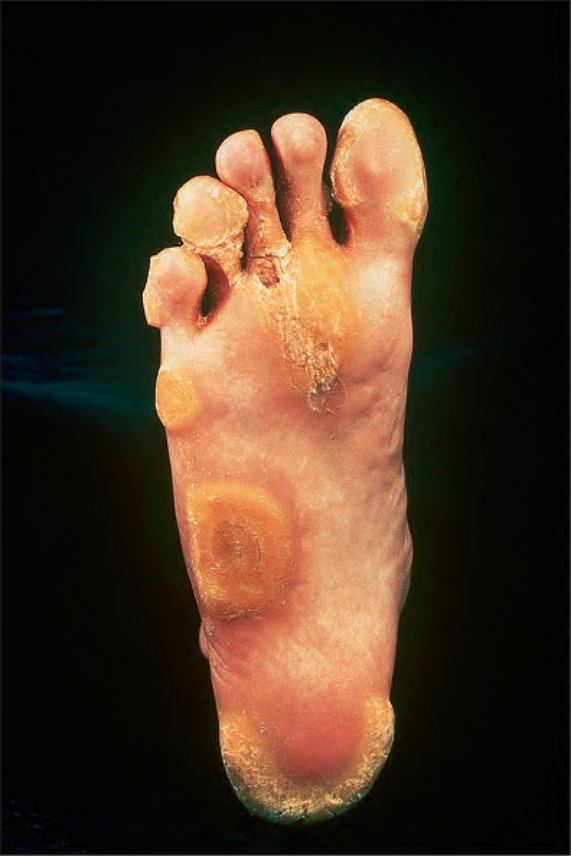
Fig. 3.118 Pachyonychia congenita: circumscribed, yellow, hyperkeratotic plaques on the soles of the feet are a common manifestation. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
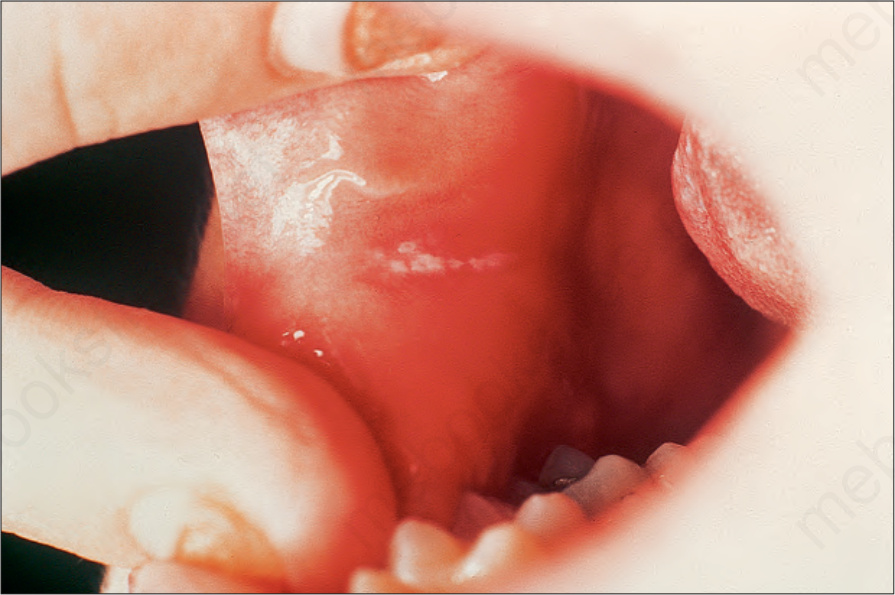
Fig. 3.119 Pachyonychia congenita: leukoplakia of the buccal mucosa is a frequent accompanying feature. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
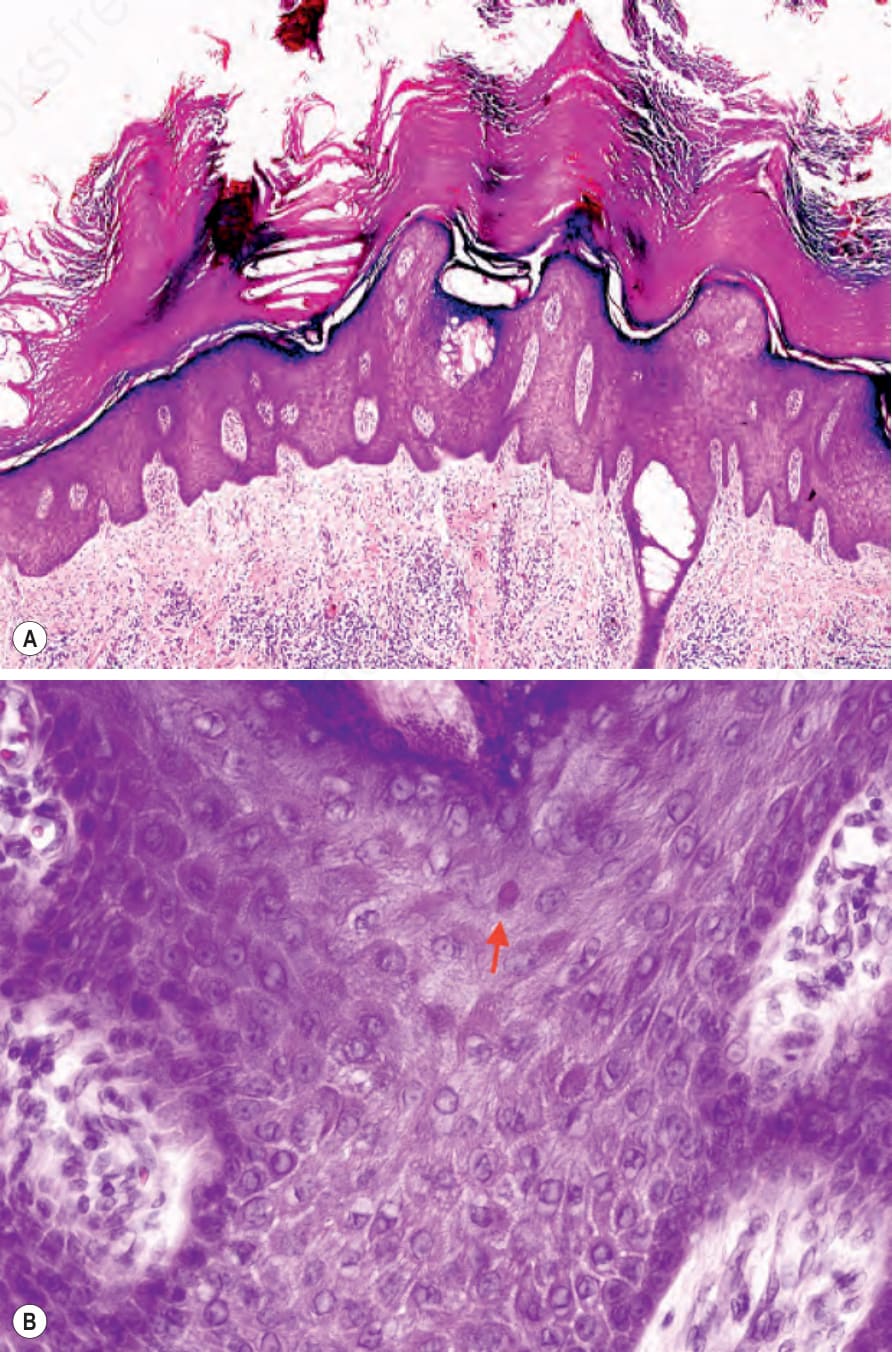
Fig. 3.120 Pachyonychia congenita: (A) volar skin showing massive hyperkeratosis, hypergranulosis, and acanthosis; (B) suprabasal keratinocytes have a characteristic pale cytoplasm and eosinophilic inclusions.
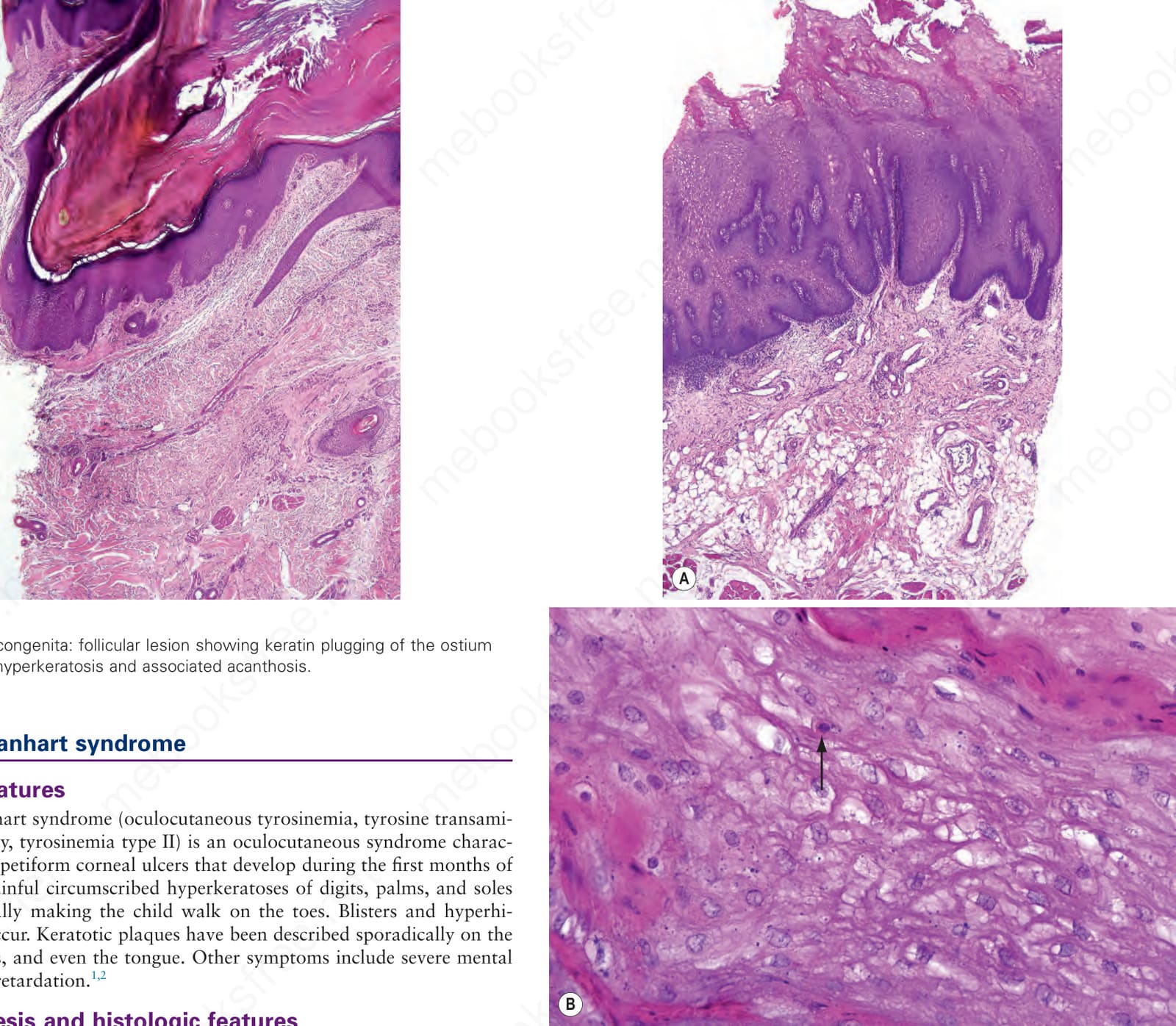
Fig. 3.121 Pachyonychia congenita: follicular lesion showing keratin plugging of the ostium with adjacent hyperkeratosis and associated acanthosis.
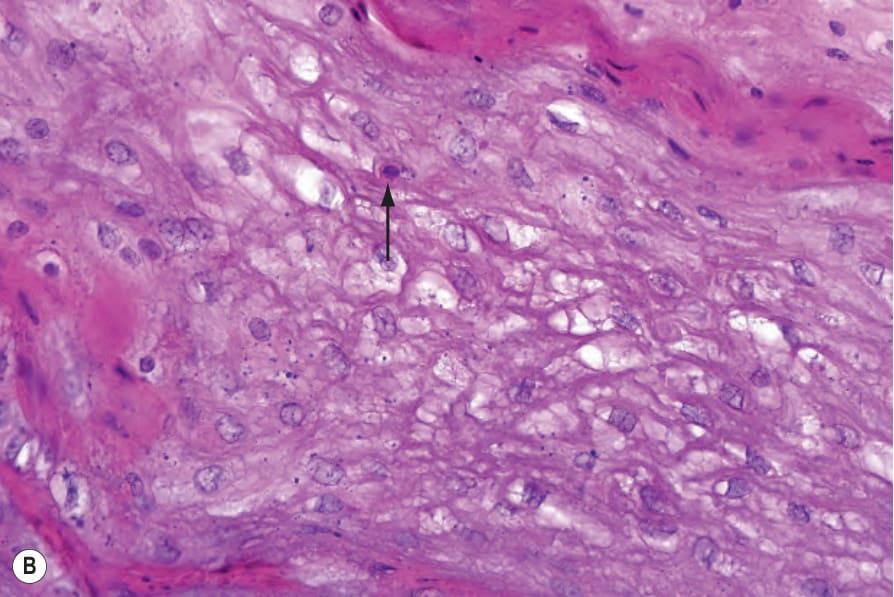
Fig. 3-122 (caption embedded in image / 圖說烘焙於圖內)
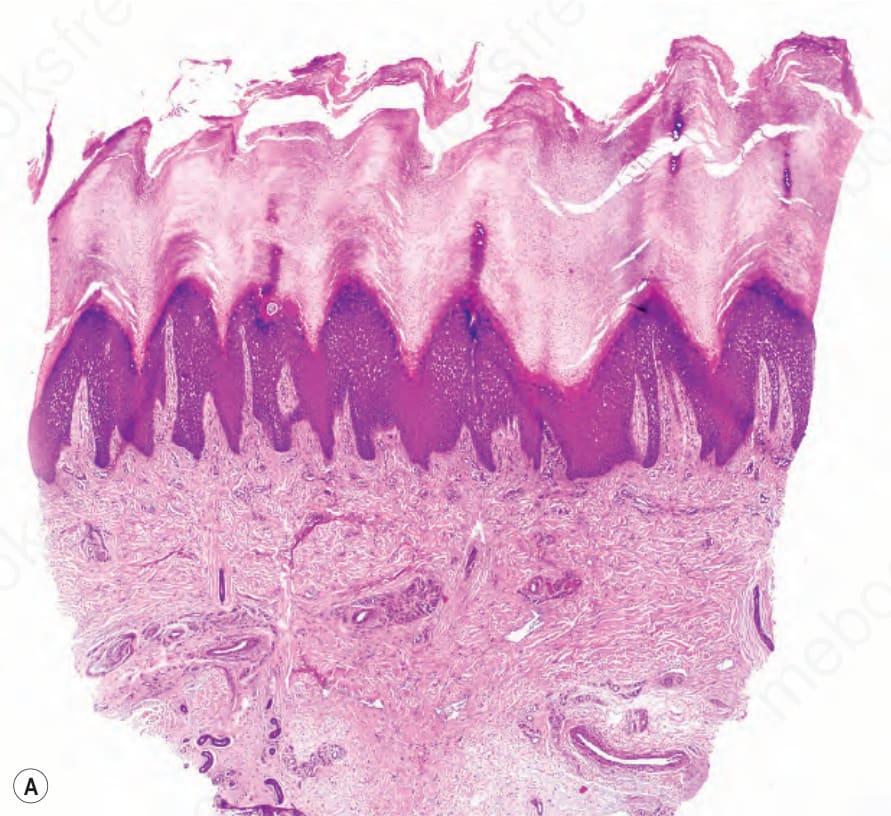
Fig. 3-117 (caption embedded in image / 圖說烘焙於圖內)