疾病定義與分類
- 掌蹠角皮症 (palmoplantar keratodermas, PPKs):一大群異質性、後天或遺傳性的角化障礙 (cornification disorders),特徵為手掌與足底過度角化 (hyperkeratosis)。
- 遺傳性 PPK 可單獨發生,或合併其他遺傳病:ichthyosis、erythrokeratoderma、epidermolysis bullosa、ectodermal dysplasia、dyskeratosis congenita。
- 症候群型可侵犯心臟、眼、聽力或神經系統;後天型可為副腫瘤性 (paraneoplastic) 或與內科疾病相關。明確診斷對治療與遺傳諮詢至關重要。
- 分類依據:遺傳方式、病灶分布、額外臨床特徵與相關異常。三大臨床類別 → 瀰漫型 (diffuse)、侷限型 (circumscribed)、點狀型 (punctate)。
遺傳性 PPK 速查(型別/遺傳/基因)
- 瀰漫型 Diffuse
- Epidermolytic PPK(Vörner-Unna-Thost;含 tonotubular 亞型、progressive PPK Greither):AD,KRT1 或 KRT9
- Non-epidermolytic PPK, Kimonis 型:AD,KRT1
- Bothnia 型:AD,AQP5
- Nagashima 型:AR,SERPINB7
- Keratolytic winter erythema:AD,基因未知
- Mal de Meleda(含 Gamborg-Nielsen):AR,SLURP1
- 侷限型 Circumscribed
- Striate PPK:AD;type 1 DSG1、type 2 DSP、type 3 KRT1
- 點狀型 Punctate
- Keratosis punctata palmoplantaris:AD;type 1 (Buschke-Fischer-Brauer) AAGAB (COL14A1)、type 2 (Spiny keratoderma)、type 3 (Marginal papular acrokeratoderma)
致病機轉/分子
- 分子缺陷可歸入數類蛋白:
- 結構蛋白:keratins
- 角化包膜 (cornified envelope):loricrin、transglutaminase
- 黏附蛋白:plakophilin、desmoplakin、desmoglein1
- 細胞間通訊:connexins、SLURP1、AAGAB
- 表皮蛋白酶:cathepsin C
臨床特徵
- 許多遺傳型屬晚發 (late onset),同一家族內表現具變異性;嚴重度受機械性壓力影響(如足部受壓)。
- 症狀:浸軟 (maceration)、水疱、惡臭、劇痛。常見併發症為多汗症 (hyperhidrosis) 與真菌感染。
組織病理特徵
- 已提出 七種主要組織學型態,特徵分別為:epidermolytic hyperkeratosis、orthohyperkeratosis 或 parakeratosis、hypergranulosis、過渡細胞 (transitional cells)、細胞間隙增寬、細胞質內嗜伊紅包涵體,或伴角化性斑塊的表皮凹陷(見 Table 3.8)。
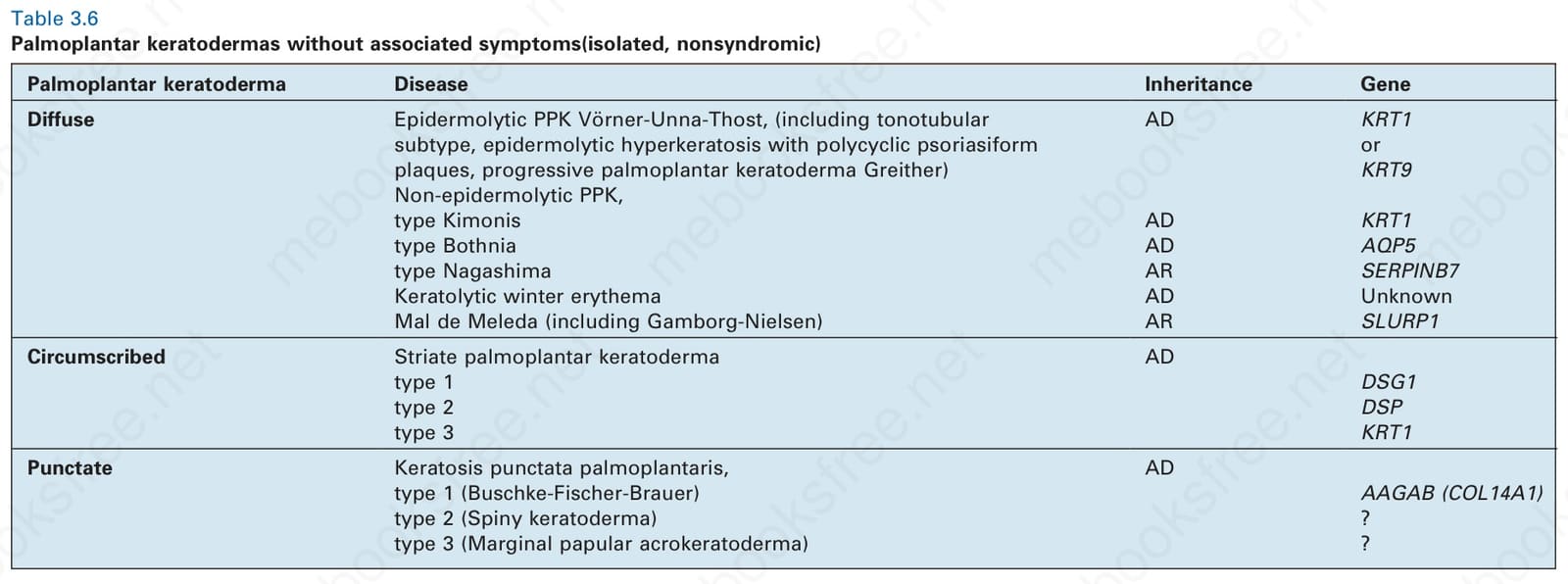
表 3-6:無相關症狀的掌蹠角皮症(孤立性、非症候群型)。
Table 3.6 Palmoplantar keratodermas without associated symptoms (isolated, nonsyndromic)
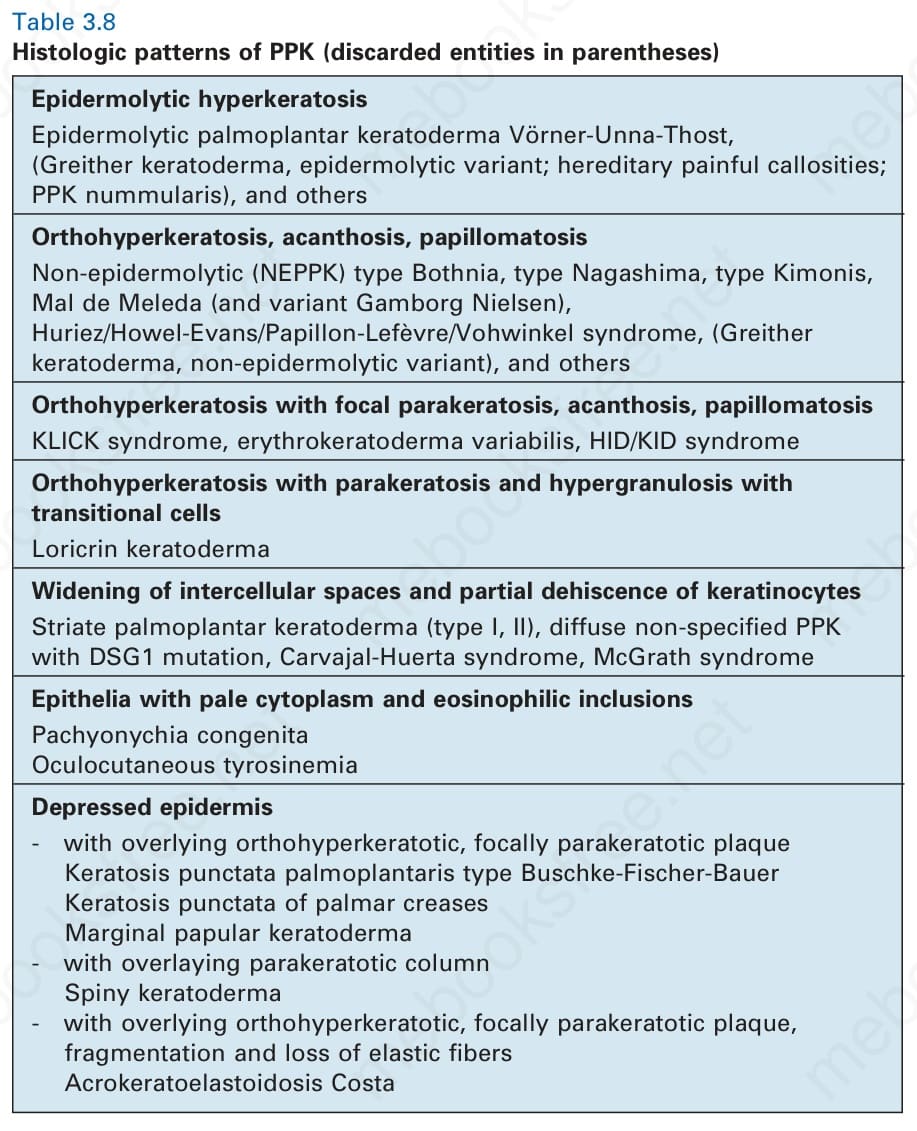
表 3-8:PPK 的組織學型態(已廢棄的疾病名置於括號內)。
Table 3.8 Histologic patterns of PPK (discarded entities in parentheses)