疾病定義與分類
- 萎縮性毛孔角化症 (keratosis pilaris atrophicans) 合併毛囊性角化過度 (follicular hyperkeratosis) 與萎縮性瘢痕 (atrophic scarring)。
- 依遺傳模式、臨床表現與相關疾病的差異,分為三型:ulerythema ophryogenes、atrophoderma vermiculata、keratosis follicularis spinulosa decalvans。
臨床特徵
- Ulerythema ophryogenes(即 keratosis pilaris atrophicans faciei, KPAF):出生時或嬰兒早期出現毛囊性丘疹伴周圍紅斑,續發眉毛外側的萎縮性瘢痕;可累及臉頰、前額、顳部、頸部,後期可致眉毛完全脫失。常為體染色體顯性 (autosomal dominant) 遺傳。
- 可伴多種遺傳症候群:Noonan syndrome、wooly hair、cardiofaciocutaneous syndrome、Cornelia de Lange syndrome、Rubinstein-Taybi syndrome、partial monosomy 18。與 Noonan syndrome 的關聯最重要——患者有可能致命的先天性肺動脈狹窄 (congenital pulmonary stenosis)。亦與 atopy 相關。
- Atrophoderma vermiculata(honeycomb atrophy;又名 ulerythema acneiforme 等):極罕見、autosomal dominant。臉頰、耳、前額出現毛囊性角化與凹陷性凹點,間夾正常皮膚呈蟲蝕狀/蜂窩狀外觀 (moth-eaten / honeycomb atrophy)。5 歲後發病;亦有沿 Blaschko lines 的單側痣樣變異。
- Keratosis follicularis spinulosa decalvans:瀰漫性萎縮性 keratosis pilaris 伴頭皮瘢痕性禿髮 (scarring alopecia);可並存 atopy、掌蹠角化過度、畏光、點狀角膜炎 (punctate keratitis)。部分為 X 連鎖隱性,因 MBTPS2 基因突變(編碼 membrane-bound transcription factor protease, site 2,定位 Xp21.13-p22.2);另有 X 連鎖顯性與體染色體顯性變異。青春期起、頭皮膿疱性的變異稱 ‘folliculitis spinulosa decalvans’。
致病機轉與組織病理特徵
- 致病機轉不明。在本病與 IFAP 中受累的 MBTPS2 可能干擾固醇 (sterol) 調控與內質網壓力反應。
- 各型共同病理:毛囊性角化過度伴管口擴張 (ostial dilatation)、皮脂腺萎縮、稀疏的毛囊周圍及/或血管周圍淋巴組織球性浸潤;可見粉刺 (comedones) 與粟丘疹 (milia);不等程度的毛囊周圍纖維化延伸至網狀真皮。
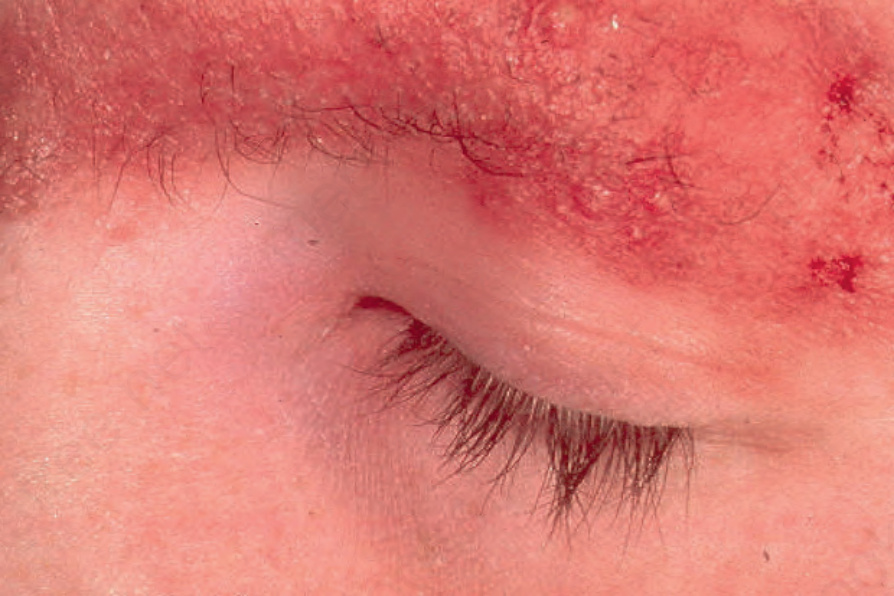
圖 3-67:Ulerythema ophryogenes:強烈紅斑伴毛囊脫失,眉毛為常見受累部位。
Fig. 3.67 Ulerythema ophryogenes: there is intense erythema with loss of follicles. The eyebrow is a commonly affected site.
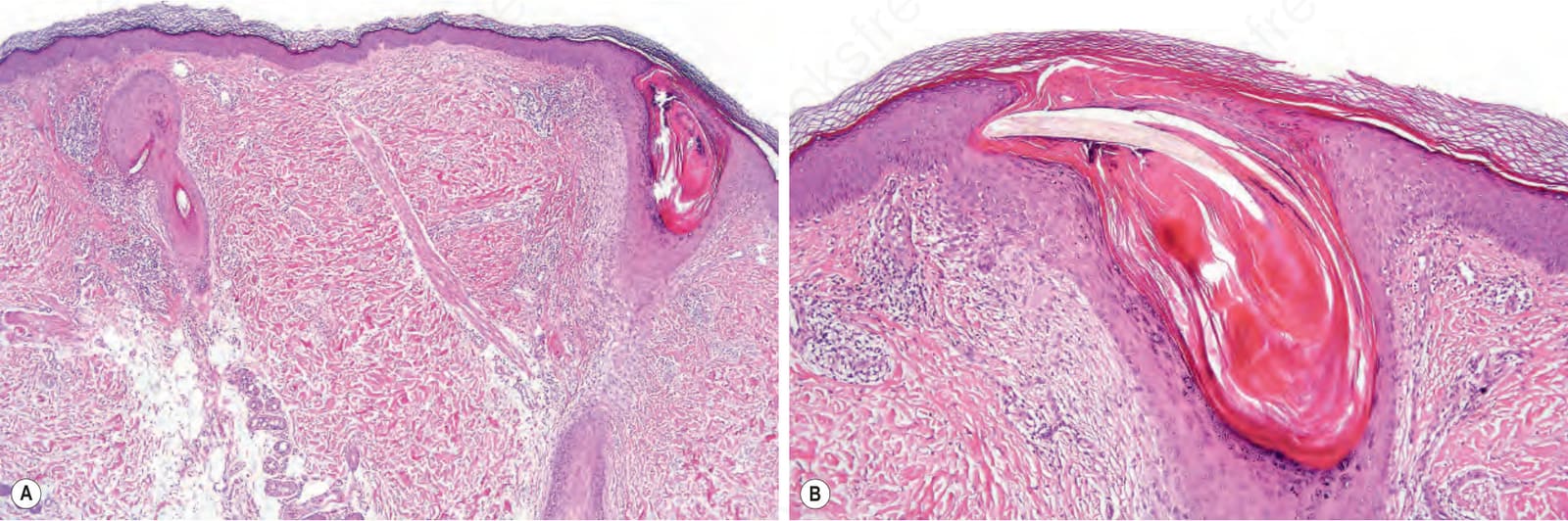
圖 3-69:萎縮性毛孔角化症:(A) 低倍可見顯著毛囊性角化過度與管口擴張;(B) 高倍可見延伸至網狀真皮的毛囊周圍纖維化。
Fig. 3.69 Keratosis pilaris atrophicans: (A) low-power view showing gross follicular hyperkeratosis and dilatation of the ostium; (B) high-power view. Note the perifollicular fibrosis extending into the reticular dermis.