臨床特徵 (Clinical Features)
- 淋巴瘤樣肉芽腫病 (lymphomatoid granulomatosis;又稱 angiocentric lymphoma、angiocentric immunoproliferative lesion;LG) 是一種罕見的結外 B 細胞增生性疾病 (extranodal B-cell proliferative disorder),伴隨反應性 T 細胞浸潤 (reactive T-cell infiltrate)、EBV 感染、免疫抑制,以及血管中心性 (angiocentric)、血管侵犯性 (angioinvasive) 與血管破壞性 (angiodestructive) 的淋巴樣浸潤。
- LG 幾乎可發生於任何年齡,從兒童晚期到老年皆有,但多數病例發生於第四至第六個十年 (fourth to sixth decades),且以男性為多 (2–3 : 1)。
致病機轉與組織學特徵 (Pathogenesis and Histologic Features)
-
lymphomatoid granulomatosis 是一種 EBV 相關的 B 細胞增生性疾病,其中佔優勢的 T 細胞族群為反應性 (reactive)。EBV 已透過原位雜合 (in situ hybridization)、Southern blot 分析及 PCR 鑑定出來,雙重標記 (double labeling) 已將病毒定位於 B 細胞內,且存在免疫球蛋白重鏈 (Ig heavy chain) 的單株性重排 (clonal rearrangements)。同一病人不同病灶內可發現不同的 B 細胞單株 (distinct clones),顯示受 EBV 感染的 B 細胞在不同部位同時發生單株擴增 (clonal expansion)。在罕見病例中,曾報告單株性 T 細胞受體基因重排 (clonal T-cell receptor gene rearrangements)。這似乎與急性及慢性 EBV 感染中偶見但已充分記載的反應性單株或寡株 T 細胞增生 (reactive clonal or oligoclonal T-cell proliferations) 現象相類似。
-
LG 主要是一種肺部疾病,超過 90% 的患者在初診時即有肺部侵犯。然而,許多其他部位也常受影響,包括皮膚 (25–50%)、腦 (26%)、腎臟 (32%) 與肝臟 (29%)。上呼吸道、胃腸道、脾臟及淋巴結的侵犯則罕見。患者通常表現為胸痛、咳嗽與呼吸困難,並伴隨發熱 (pyrexia)、倦怠 (malaise) 與體重減輕。胸部 X 光片常顯示雙側圓形結節狀陰影 (bilateral round nodular opacities),較少見的是瀰漫性絨毛狀浸潤 (diffuse fluffy infiltration)。中樞神經系統 (CNS) 侵犯與不良預後相關,可能表現為意識混亂 (confusion)、運動失調 (ataxia)、癲癇 (epilepsy)、上運動神經元徵象 (upper motor neuron signs)、顱神經麻痺 (cranial nerve palsies) 與周邊神經病變 (peripheral neuropathies)。肌痛 (myalgia)、關節痛 (arthralgia) 與胃腸道症狀則罕見。
-
本病與潛在的免疫缺陷 (underlying immunodeficiency) 有關。LG 的風險在異體器官移植 (allogeneic organ transplant) 受者,以及罹患 Wiskott-Aldrich syndrome、HIV 感染與 X-linked lymphoproliferative syndrome 的患者中升高。LG 亦曾被描述與 CLL 及 angioimmunoblastic T-cell lymphoma 相關,這些血液惡性腫瘤皆為眾所周知與 T 細胞功能缺陷有關者。此外,實驗室分析通常顯示,即使在沒有易感性免疫缺陷病史的 lymphomatoid granulomatosis 患者中,也有免疫功能下降的證據。常出現並導致組織壞死的血管變化,被認為源自 EBV 相關之趨化因子 IP-10 與 Mig 的上調 (up-regulation),這些趨化因子已知會損傷血管內皮細胞 (vascular endothelial cells) 並促進 T 細胞黏附。
-
當有皮膚侵犯時,結節 (nodules) 與丘疹 (papules) 的切片提供最具診斷性的組織學表現,因為斑塊樣病灶 (plaquelike lesions) 的變化常為非特異性。LG 的特徵為血管周圍 (perivascular) 之血管破壞性,以及附屬器周圍 (periadnexal) 之淺層與深層多形性淋巴樣浸潤 (polymorphous lymphoid infiltrate),可延伸至皮下,並因脂肪壞死 (fat necrosis) 而局部形成肉芽腫 (granulomas) (Figs 29.228–29.231)。儘管病名如此,本病一般不見其他肉芽腫。以小淋巴細胞為主,並伴隨漿細胞 (plasma cells)、組織細胞 (histiocytes) 與大型轉化淋巴樣細胞 (large transformed lymphoid cells)。某些情況下,小淋巴細胞具有不規則或略為增大的細胞核,部分作者稱之為「非典型 (atypia)」,但並無核深染 (nuclear hyperchromasia) 或惡性細胞學特徵。大型轉化細胞常類似免疫母細胞 (immunoblasts),但有些細胞較大,具有豐富的細胞質、多形性囊泡狀核 (pleomorphic vesicular nuclei) 與明顯核仁 (prominent nucleoli)。可見多核形 (multinucleate forms),部分類似 Hodgkin cells,但不應出現典型的 Reed-Sternberg cells。嗜中性球 (neutrophils) 與嗜酸性球 (eosinophils) 一般不顯著或缺如。通常可見血管壁 (動脈與靜脈皆然) 的浸潤,且可能有纖維素樣壞死 (fibrinoid necrosis),伴隨周圍實質廣泛的凝固性壞死 (coagulative necrosis)。
免疫組化與特殊染色 (Immunohistochemistry & Special Stains)
- 透過免疫組織化學,背景淋巴細胞族群為 CD3+ T 細胞,主要為 CD4+ 輔助型 (helper subtype)。然而,大型非典型轉化細胞為 B 細胞,具 CD20+/CD79a+ 免疫表型。CD30 常為陽性,但 CD15、CD56 與 CD57 一律為陰性。不定數量的細胞顯示 EBV 證據,而以 EBV 編碼 RNA (EBERs) 的原位雜合 (in situ hybridization) 比以潛伏膜蛋白 1 (latent membrane protein 1, LMP1) 的免疫組織化學更能顯示之。EBV 在皮膚病灶中比在肺部病灶中更難顯示。輕鏈限制 (light chain restriction) 僅罕見地被辨識出,且僅見於呈漿細胞樣分化 (plasmacytoid differentiation) 之細胞的細胞質中。血管侵犯性成分通常也包含反應性 CD4+ T 輔助細胞。
分級 (Grading)
- LG 的預後在某種程度上反映其組織學分級 (histologic grade)。具有更明顯淋巴瘤外觀的腫瘤行為更具侵襲性。分級與 EBV 陽性 B 細胞相對於反應性背景的比例有關。
- Grade I 病灶高度多形性,且不顯示淋巴細胞核非典型 (lymphocytic nuclear atypia)。母細胞 (blast cells) 不顯著,常僅在免疫組織化學染色切片中可偵測。壞死缺如或非常局部。EBV+ 細胞非常稀少 (以 in situ hybridization 計每高倍視野 <5 個)。
- Grade II 病灶顯示淋巴細胞細胞學非典型 (lymphocytic cytological atypia),大型轉化細胞數量較多且可形成小群聚。壞死常見。EBV+ 細胞數為每高倍視野 5–20 個。
- Grade III 病灶為明確的淋巴瘤性 (frankly lymphomatous),但仍可見多形性發炎背景浸潤。大型非典型轉化細胞易於辨識,並可形成較大的聚集。EBV+ 細胞非常顯著 (每高倍視野 >50 個),並可呈現為融合成片 (confluent sheets)。當出現一致性的 EBV+ 母細胞族群且無發炎背景時,則不再能維持 LG 的診斷,該病灶應視為 diffuse large B-cell lymphoma。
鑑別診斷 (Differential Diagnosis)
- Wegener granulomatosis 在臨床上與 LG 的不同處在於其上呼吸道侵犯,以及伴隨肉芽腫性發炎 (granulomatous inflammation) 的壞死性血管炎 (necrotizing vasculitis)。可能與 LG 混淆的淋巴增生性疾病 (lymphoproliferative disorders) 通常也呈現部分血管中心性/血管破壞性 (angiocentric/angiodestructive) 的生長型態,包括 extranodal NK/T-cell lymphoma、Epstein-Barr virus-positive mucocutaneous ulcer (EBV+ MCU) 與 EBV-positive diffuse large B-cell lymphoma。肺部侵犯在 extranodal NK/T-cell lymphoma 中不常見,且雖然也與 EBV 感染相關,但其特徵為 CD3–、CD20–、CD3ε+、CD56+ 表型。EBV+ MCU 與 EBV-positive diffuse large B-cell lymphoma 與 LG 譜系的某些部分有相當大的病理重疊,必須將臨床特徵納入考量方能達成正確診斷。EBV+ MCU 始終為局部性,不形成腫塊性病灶 (mass lesion)。在病理上,它是一個界限分明的病灶 (circumscribed lesion),在浸潤的基部與側緣具有局限性的小 T 細胞邊緣 (confining rim)。要做出 LG 的診斷必須見到特徵性的肺部變化,且與 EBV-positive diffuse large B-cell lymphoma 不同,淋巴結侵犯通常缺如。
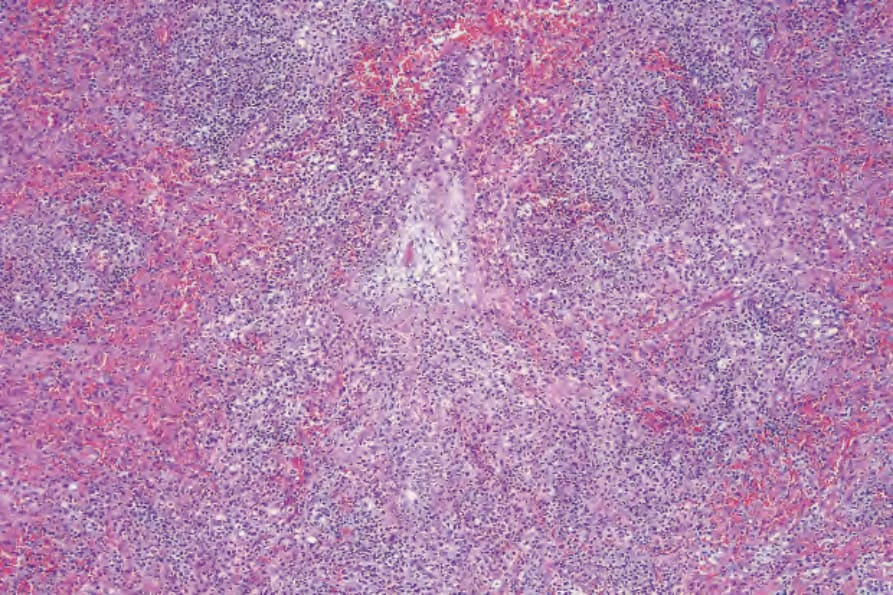
圖 29-228:淋巴瘤樣肉芽腫病:可見多結節性腫瘤細胞浸潤 (Lymphomatoid granulomatosis; multinodular tumor cell infiltrate)。
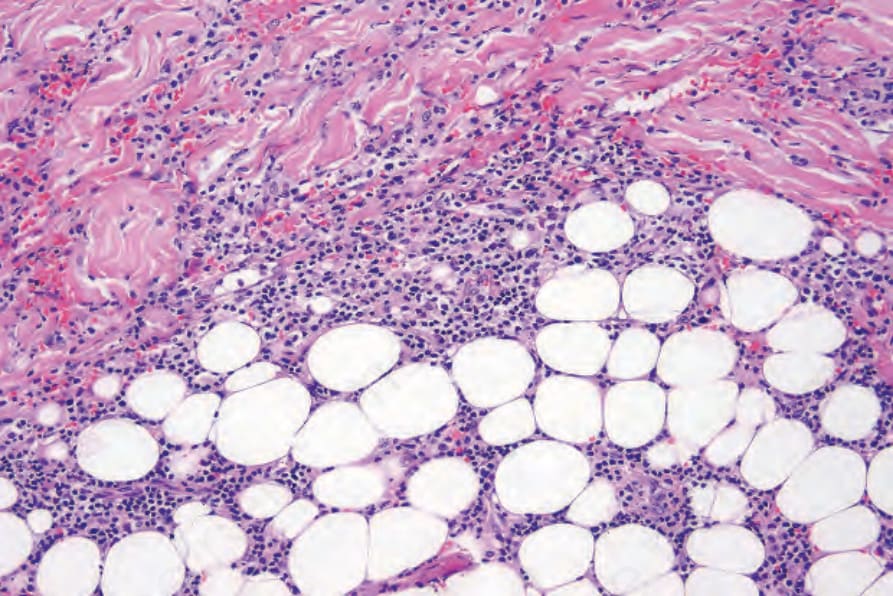
圖 29-229:淋巴瘤樣肉芽腫病:浸潤延伸至皮下脂肪 (Lymphomatoid granulomatosis; infiltrate extends into the subcutaneous fat)。
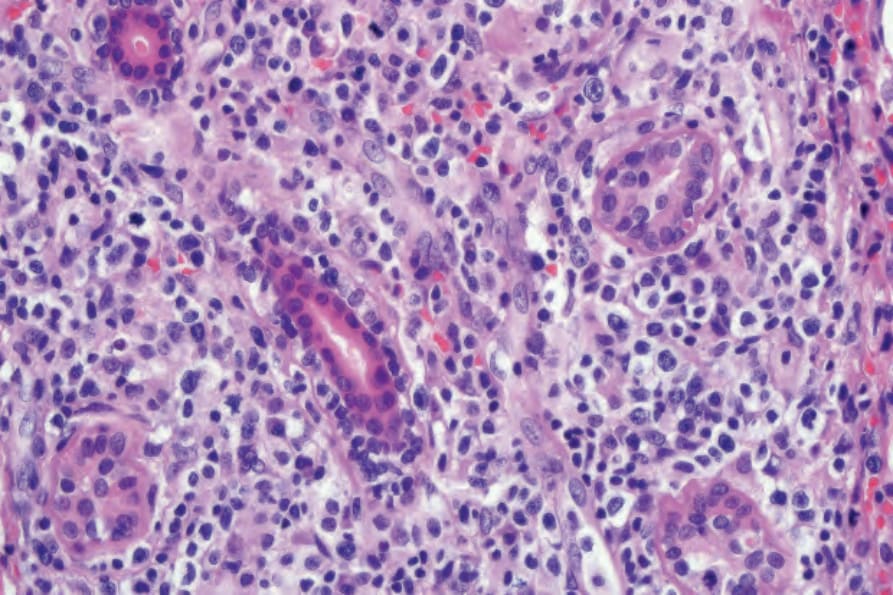
圖 29-230:淋巴瘤樣肉芽腫病:真皮高倍視野顯示非典型淋巴樣細胞 (Lymphomatoid granulomatosis; high-power view of dermis showing atypical lymphoid cells)。
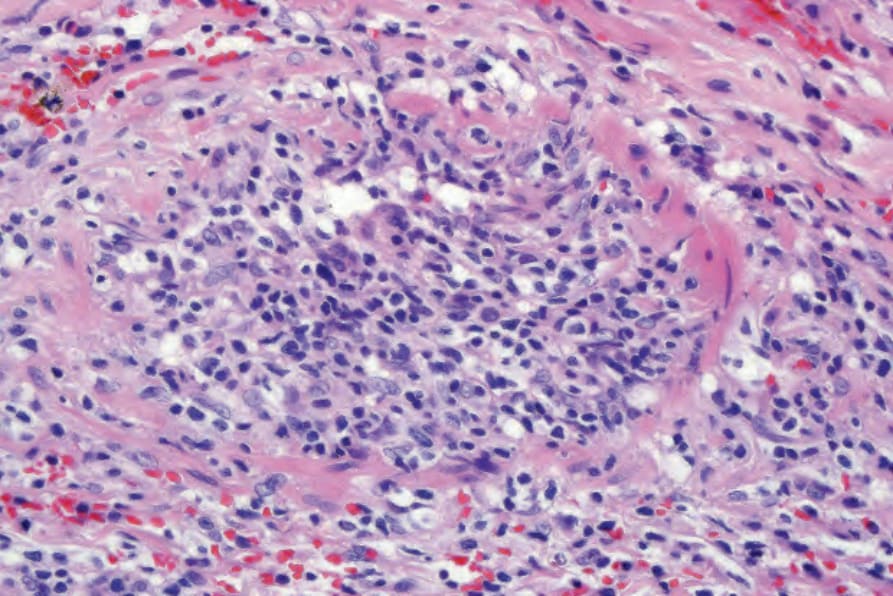
圖 29-231:淋巴瘤樣肉芽腫病:可見顯著的血管侵犯 (Lymphomatoid granulomatosis; striking vascular involvement)。
漿細胞骨髓瘤的皮膚表現與原發性皮膚漿細胞瘤 (Cutaneous Manifestations of Plasma Cell Myeloma and Primary Cutaneous Plasmacytoma)
臨床特徵 (Clinical Features)
-
除了腫瘤細胞特異性浸潤所致的表現外,多種皮膚病況與漿細胞腫瘤 (plasma cell neoplasms) 相關。許多與單株免疫球蛋白 (monoclonal immunoglobulin) 或其片段之一的沉積有關,這些包括澱粉樣變性 (amyloidosis) 與冷球蛋白血症 (cryoglobulinemia)。其他則是本身獨立的特定疾病,但顯示與漿細胞異常增生 (plasma cell dyscrasias) 相關,這些包括嗜中性球皮膚病 (neutrophilic dermatoses)、白血球碎裂性血管炎 (leukocytoclastic vasculitis)、硬化性黏液水腫 (scleromyxedema)、硬皮病 (scleroderma)、角質下膿疱性皮膚病 (subcorneal pustular dermatoses)、壞死性黃色肉芽腫 (necrobiotic xanthogranuloma)、角質下膿疱性皮膚病 (subcorneal pustular dermatosis) 與 POEMS syndrome。此外,還有一群雜項非特異性病況,包括搔癢 (pruritus)、感染與藥物反應。關於這些疾病更詳細的描述可在本書他處或回顧文章中找到。本節討論由腫瘤過程造成的皮膚浸潤之後果。
-
在 plasma cell myeloma 中皮膚侵犯相對罕見,初診時發生於 3–4% 的病例,復發或疾病進展時約發生於 5% 的病例。皮膚侵犯最常代表 plasma cell myeloma 或廣泛髓外漿細胞瘤 (extramedullary plasmacytoma) 的終末期表現,但偶爾它會先於本病的其他表現出現。它可能因血行性散播 (hematogenous spread),或由其下方骨性沉積物的直接擴展而發展。骨髓瘤的髓外散播 (extramedullary dissemination),包括散播至皮膚部位,通常與極差的預後相關,多數患者於發生後數月內死亡。
組織學特徵 (Histologic Features)
- 原發性皮膚漿細胞瘤 (primary cutaneous plasmacytoma) 與轉移性骨髓瘤 (metastatic myeloma) 的組織學特徵相似。浸潤可為結節狀 (nodular) 或瀰漫性 (diffuse),通常範圍廣泛,常侵犯皮下脂肪 (Figs 29.232 and 29.233)。常見一個清楚的無浸潤帶 (grenz zone)。在繼發性皮膚侵犯的病例中,曾描述間質性生長型態 (interstitial pattern of growth)。雖然在某些分化良好的例子中,浸潤的漿細胞本質很明顯,具有特徵性的鐘面狀核 (clock-face nuclei),但更常見的是腫瘤細胞呈非典型,具有明顯成角且常為塑形 (molded) 的細胞質邊界,多形性核含明顯核仁,以及顆粒狀嗜酸性細胞質 (granular eosinophilic cytoplasm)。常出現雙核 (binucleate) 與多核 (multinucleate) 形,在分化較差的病灶中有絲分裂象 (mitotic figures) 可能豐富。胞質內包涵體 (Russell bodies) 與核內包涵體 (Dutcher bodies) 雖然通常不顯著,但有時可出現。極少數情況下,可在腫瘤相關組織細胞的細胞質中辨識出嗜酸性菱形或針狀晶體 (eosinophilic rhomboidal or needle-shaped crystals)(晶體儲存性組織細胞增生症,crystal-storing histiocytosis),此情況更常與 plasma cell myeloma 相關 (Fig. 29.234)。亦曾報告一例繼發性黏蛋白沉積 (secondary mucinosis)。
Fig. 29.228 Lymphomatoid granulomatosis: there is a multinodular tumor cell infiltrate.
Fig. 29.229 Lymphomatoid granulomatosis: the infiltrate extends into the subcutaneous fat.
圖 29-230:淋巴瘤樣肉芽腫病 (lymphomatoid granulomatosis):真皮高倍視野顯示非典型淋巴樣細胞。
Fig. 29.230 Lymphomatoid granulomatosis: high-power view of dermis showing atypical lymphoid cells.
Fig. 29.231 Lymphomatoid granulomatosis: there is striking vascular involvement.
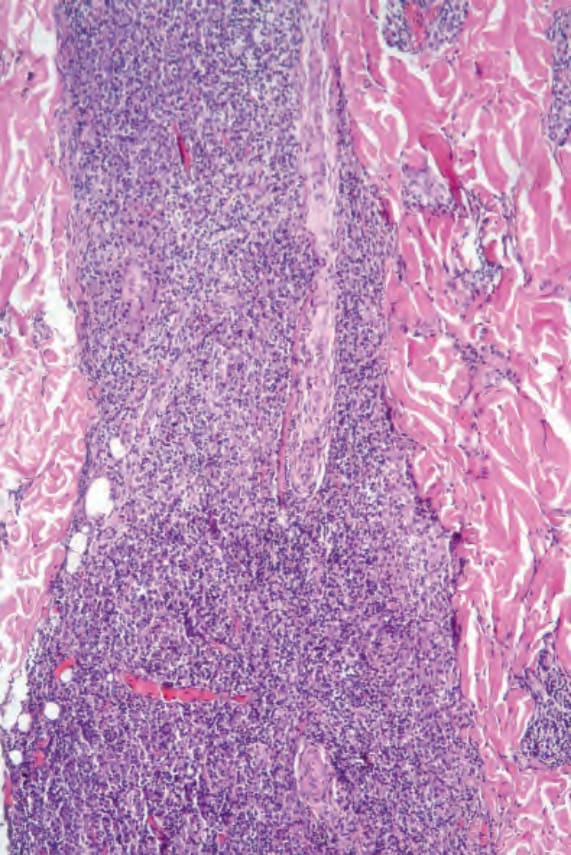
Fig. 29.232 Plasmacytoma: there is a dense dermal infiltrate. By courtesy of J. Cohen, MD, Dermatopathology Laboratory, Tucson, Arizona, USA.
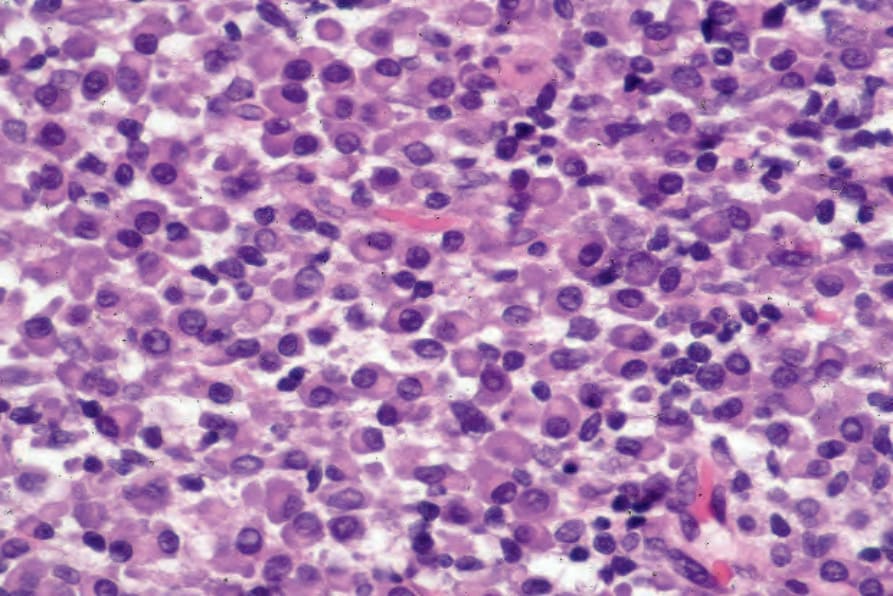
圖 29-233:漿細胞瘤 (plasmacytoma):浸潤幾乎全由純漿細胞群組成。
Fig. 29.233 Plasmacytoma: the infiltrate consists of an almost pure plasma cell population. By courtesy of J. Cohen, MD, Dermatopathology Laboratory, Tucson, Arizona, USA.
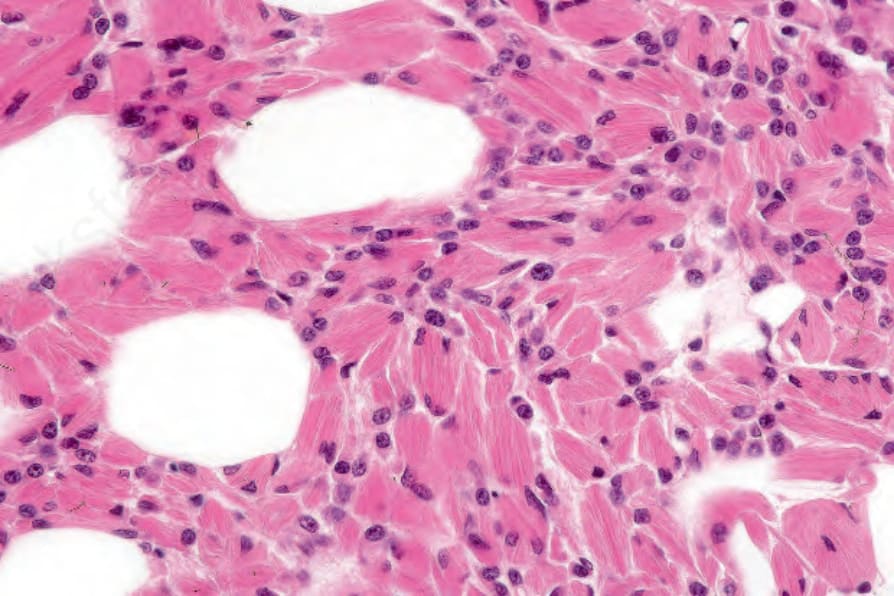
Fig. 29.234 Multiple myeloma: histiocytes containing needle-shaped crystals are evident – so-called crystal-storing histiocytosis. By courtesy of G. Pinkus, MD, Brigham and Women’s Hospital and Harvard Medical School, Boston, USA.
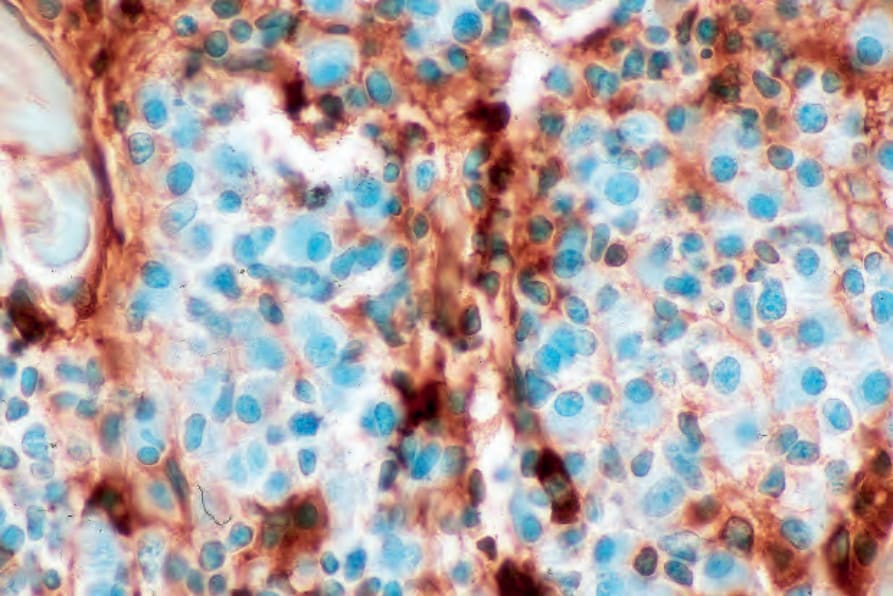
圖 29-237:漿細胞瘤 (plasmacytoma):lambda light chain 缺如。
Fig. 29.237 Plasmacytoma: lambda light chain is absent. By courtesy of J. Cohen, MD, Dermatopathology Laboratory, Tucson, Arizona, USA.
-
骨髓瘤沉積物 (myelomatous deposits) 表現為直徑 1–5 cm 的單發、或更常為多發的膚色、紅色或紫紅色 (violaceous) 結節,依頻率遞減順序影響軀幹、四肢與臉部。
-
primary cutaneous plasmacytomas 屬於骨外 (髓外) 漿細胞瘤 (extraosseus [extramedullary] plasmacytomas) 群。這些被定義為發生於骨以外組織的漿細胞腫瘤,佔所有漿細胞腫瘤的 3–5%。八成的病例發生於上呼吸道,但也記載有發生於胃腸道、淋巴結、膀胱、腦、甲狀腺、睪丸、腮腺與皮膚者。因此,primary cutaneous plasmacytomas 屬例外,英語文獻中僅記載少數真正的病例。病灶可為單發或更常為多發,依頻率遞減順序出現於臉部、軀幹、四肢、頭皮與手指。在多數報告中,它們被描述為紅色至紫色 (red to purple) 的結節或斑塊。中年與老年人為主要受影響者 (平均 60 歲;範圍 22–88 歲),且有男性偏好 (4 : 1)。罕見病例發生於實體器官移植受者。單一腫瘤的患者有時可能預後較佳,但多發病灶常與淋巴結及內臟侵犯,或 plasma cell myeloma 的發展及高死亡率相關。整體死亡率至少為 40%。然而,偶爾可見長期無病存活 (long-term disease-free survival)。
免疫組化 (Immunohistochemistry)
- 透過免疫組織化學,漿細胞表現 CD38、CD79a 與 CD138 (Fig. 29.235)。它們通常 CD19 陰性,且常缺乏 CD20 與共同白血球抗原 (common leukocyte antigen)。可見單型性胞質內免疫球蛋白輕鏈 (monotypic cytoplasmic Ig light chain) (Figs 29.236 and 29.237)。可能表現 CD56 與 cyclin D1,偶爾對 EMA、HMB-45 與細胞角蛋白 (cytokeratin) 呈陽性。
鑑別診斷 (Differential Diagnosis)
- 本病分化不良型 (poorly differentiated forms) 與 plasmablastic lymphoma 的區別於 plasmablastic lymphoma 一節中討論。分化良好的腫瘤有時會被誤認為富含漿細胞的病況,如 syphilis 或 Borrelia 感染。若存在顯著的背景淋巴細胞族群,則可能與 primary cutaneous marginal zone B-cell lymphoma/immunocytoma 或 lymphoplasmacytic lymphoma 混淆。此時的區分有賴於以形態學 (marginal zone cells)、免疫組織化學 (CD20) 或流式細胞術 (flow cytometry) 證明腫瘤性淋巴細胞成分。對於分化不良的例子,鑑別診斷可包括高惡性度淋巴瘤 (high-grade lymphoma)、癌 (carcinoma) 或黑色素瘤 (melanoma)。在此類情況下,免疫組織化學抗體組合需要擴充。
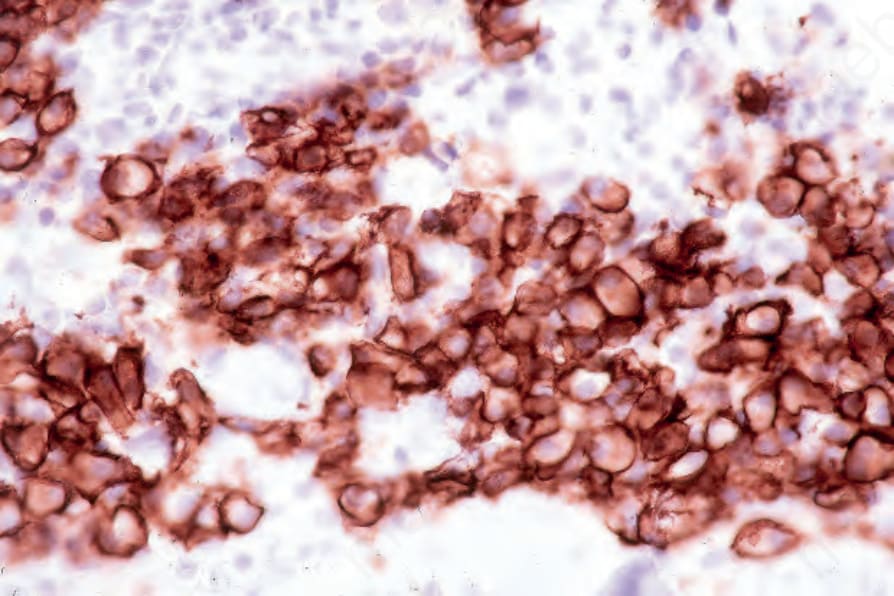
Fig. 29.235 Plasmacytoma: the tumor cells express CD138. By courtesy of J. Cohen, MD, Dermatopathology Laboratory, Tucson, Arizona, USA.
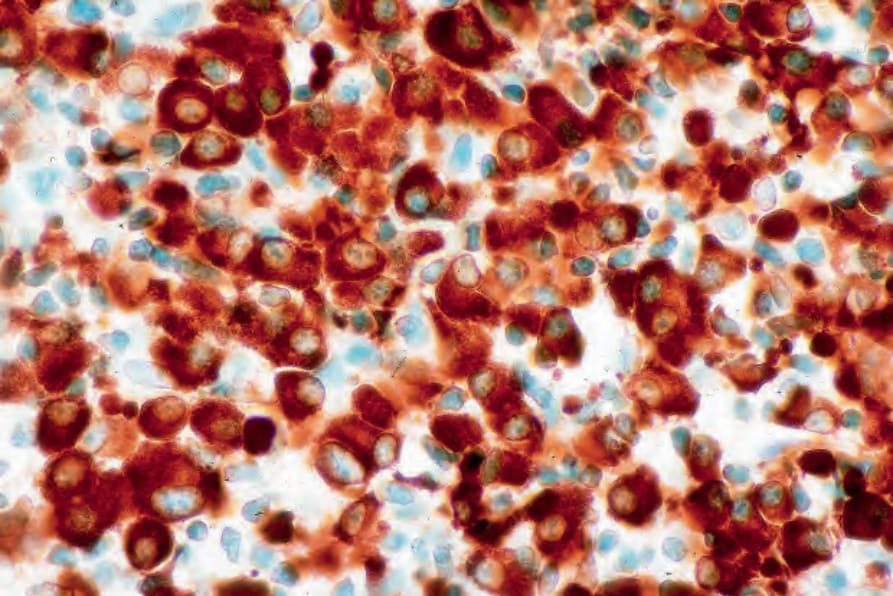
圖 29-236:漿細胞瘤 (plasmacytoma):呈現一致的 kappa light chain 表現。
Fig. 29.236 Plasmacytoma: there is uniform kappa light chain expression. By courtesy of J. Cohen, MD, Dermatopathology Laboratory, Tucson, Arizona, USA.