Lymphomatoid granulomatosis
Lymphomatoid granulomatosis
Clinical features Lymphomatoid granulomatosis (angiocentric lymphoma, angiocentric immunoproliferative lesion) (LG) is a rare extranodal B-cell proliferative disorder associated with a reactive T-cell infiltrate, EBV infection, immunosuppression, and an angiocentric, angioinvasive, and angiodestructive lymphoid infiltrate.1–7 LG may present at virtually any age, from late childhood to old age, but most cases occur in the fourth to sixth decades with male predominance (2–3 : 1).2–5,8–10
Pathogenesis and histologic features Lymphomatoid granulomatosis is an EBV-associated B-cell proliferative disorder in which the T-cell predominant population is reactive.15 EBV has been identified by in situ hybridization, Southern blot analysis, and PCR and double labeling has identified the virus within B cells,18–23 and clonal rearrangements of the Ig heavy chain are present.12,23,24 Distinct clones of B cells may be found in different lesions within the same patient, indicating that clonal expansion of EBV-infected B cells takes place simultaneously at different sites.12–15 In rare cases, clonal T-cell receptor gene rearrangements have been reported. This seems analogous to the occasional but well-documented finding of reactive clonal or oligoclonal T-cell proliferations in association with acute and chronic EBV infection.10,12,25,26
LG is primarily a pulmonary disease, with lung involvement being present in >90% of patients at presentation. However, a number of other sites are commonly affected including the skin (25–50%), the brain (26%), kidneys (32%), and liver (29%). Involvement of the upper respiratory and gastrointestinal tracts, spleen, and lymph nodes is rare.2–5,7 Patients usually present with chest pain, cough, and dyspnea in addition to pyrexia, malaise, and weight loss.2–4 The chest radiograph commonly shows bilateral round nodular opacities or, less commonly, diffuse fluffy infiltration.11 CNS involvement is associated with poor prognosis and may manifest as confusion, ataxia, epilepsy, upper motor neuron signs, cranial nerve palsies, and peripheral neuropathies.7 Myalgia, arthralgia, and gastrointestinal symptoms are rare.
There is an association with underlying immunodeficiency.24,27 The risk of LG is increased in allogeneic organ transplant recipients and in patients with Wiskott-Aldrich syndrome, HIV infection, and X-linked lymphoproliferative syndrome.9,27–31 LG has also been described in association with CLL and angioimmunoblasic T-cell lymphoma, hematological malignancies well known to be associated with defects in T-cell function.32 Moreover, laboratory analysis usually reveals evidence of reduced immune function in patients presenting with lymphomatoid granulomatosis in the absence of a history of predisposing immunodeficiency.15,33 The vascular changes that are often present and lead to tissue necrosis are believed to result from EBV-related up-regulation of the chemokines IP-10 and Mig, which are known to damage vascular endothelial cells and to promote adhesion of T cells.12,34
1473 Lymphomatoid granulomatosis
Biopsies of nodules and papules provide the most diagnostic histology when there is cutaneous involvement, as the changes in plaquelike lesions are often non-specific.12 LG is characterized by a perivascular angiodestructive and periadnexal superficial and deep polymorphous lymphoid infiltrate that may extend into the subcutis with focal formation of granulomas due to fat necrosis (Figs 29.228–29.231).1–4 Granulomas are not otherwise seen in the disease despite its name. Small lymphocytes predominate and are accompanied by plasma cells, histiocytes, and large transformed lymphoid cells. In some instances, the small lymphocytes have irregular or slightly enlarged nuclei, and this has been referred to as ‘atypia’ by some authors, although there is no nuclear hyperchromasia or cytological features of malignancy.12 The large transformed cells often resemble immunoblasts, but some are large with abundant cytoplasm and pleomorphic vesicular nuclei and prominent nucleoli. Multinucleate forms may be seen and some resemble Hodgkin cells, although classic Reed-Sternberg cells should not be present.1 Neutrophils and eosinophils are generally inconspicuous or absent.1 Infiltration of blood vessel walls (both arteries and veins) is usually seen, and there may be fibrinoid necrosis with widespread coagulative necrosis of the surrounding parenchyma.2–4
immunophenotype.1,4 CD30 is often present but CD15, CD56, and CD57 are invariably negative.4,15,23,24,35 A variable number shows evidence of EBV, and this is better demonstrated by in situ hybridization for EBV-encoded RNA (EBERs) than with immunohistochemistry for latent membrane protein 1 (LMP1). EBV is harder to demonstrate in cutaneous than in pulmonary lesions.22 Light chain restriction is only rarely identified, and then in the cytoplasm of cells displaying plasmacytoid differentiation.1 The angioinvasive component also commonly comprises reactive CD4+ T-helper cells.
By immunohistochemistry, the background population of lymphocytes is CD3+ T cells, predominantly of the CD4+ helper subtype. The large atypical transformed cells, however, are B cells with a CD20+/CD79a+
Grading The prognosis of LG is, to some extent, a reflection of the histologic grade. Tumors with a more overt lymphomatous appearance have a more aggressive behavior. Grade is related to the proportion of EBV-positive B cells relative to the reactive background.1,2,4,15,24,36
• Grade I lesions are highly polymorphous and show no lymphocytic nuclear atypia. Blast cells are inconspicuous and often only detected in immunohistochemically stained sections. Necrosis is absent or very focal. EBV+ cells are very sparse (<5 per HPF with in situ hybridization).
• Grade II lesions show lymphocytic cytological atypia, and large transformed cells are more numerous and may form small clusters. Necrosis is common. EBV+ cells number 5–20 per HPF.
1474 Cutaneous lymphoproliferative diseases and related disorders
• Grade III lesions are frankly lymphomatous, although a polymorphous inflammatory background infiltrate is seen. Large atypical transformed cells are readily identifiable and may form larger aggregates. EBV+ cells are very conspicuous (>50 per HPF) and may present as confluent sheets. When a uniform population of EBV+ blasts is present with no inflammatory background then a diagnosis of LG is no longer tenable, and the lesion should be regarded as diffuse large B-cell lymphoma.
Differential diagnosis Wegener granulomatosis differs clinically from LG by its upper respiratory tract involvement and by the presence of necrotizing vasculitis accompanied by granulomatous inflammation. Lymphoproliferative disorders which can be confused with LG usually also display a partial angiocentric/angiodestructive growth pattern and include extranodal NK/T-cell lymphoma, Epstein-Barr virus-positive mucocutaneous ulcer (EBV+ MCU) and EBV-positive diffuse large B-cell lymphoma. Pulmonary involvement is uncommon in extranodal NK/T-cell lymphoma, and although also associated with EBV infection, is characterized by a CD3–, CD20–, CD3ε+, CD56+ phenotype. EBV+ MCU and EBV-positive diffuse large B-cell lymphoma show considerable pathological overlaps with parts of the LG spectrum, and clinical features must be taken into account in order to arrive at the correct diagnosis. EBV+ MCU is always localized without formation of a mass lesion. Pathologically, it is a circumscribed lesion with a confining rim of small T cells at the base and sides of the infiltrate. Characteristic lung changes should be seen in order to make a diagnosis of LG and, unlike EBV-positive diffuse large B-cell lymphoma, lymph node involvement is typically absent.
Cutaneous manifestations of plasma cell myeloma and primary cutaneous plasmacytoma
Clinical features A wide variety of skin conditions are associated with plasma cell neoplasms, apart from the manifestations of specific infiltration by tumor cells. Many are associated with deposition of monoclonal immunoglobulin or one of its fragments. These include amyloidosis and cryoglobulinemia. Others are specific diseases in their own right but show an association with plasma cell dyscrasias. These include neutrophilic dermatoses, leukocytoclastic vasculitis, scleromyxedema, scleroderma, subcorneal pustular dermatoses, necrobiotic xanthogranuloma, subcorneal pustular dermatosis, and POEMS syndrome.1,2 In addition, there is a miscellaneous group of non-specific conditions that includes pruritus, infections, and drug reactions.1,2 A more detailed account of these disorders can be found elsewhere in this book or in review articles.1,2 This section deals with the consequences of cutaneous infiltration by the neoplastic process.
solitary or more often multiple, present on the face, trunk, limbs, scalp, and finger in decreasing order of frequency.8,14–20 In most reports, they have been described as red to purple nodules or plaques. The middle aged and elderly are predominantly affected (mean, 60 years; range, 22–88 years), and there is a predilection for males (4 : 1).13 Rare cases occur in solid organ transplant recipients.2,21,22 Patients with single tumors may sometimes have a better prognosis, but multiple lesions are commonly associated with nodal and visceral involvement or development of plasma cell myeloma and a high mortality.13,15,23 The overall death rate is at least 40%.17 Occasionally, however, long-term disease-free survival is seen.13
Skin involvement is relatively rare in plasma cell myeloma, occurring in 3–4% of cases at presentation and approximately in 5% of cases at relapse or disease progression.3 Cutaneous involvement most often represents terminal expression of plasma cell myeloma or widespread extramedullary plasmacytoma, but occasionally it precedes other manifestations of the disease.2–8 It may develop as a consequence of hematogenous spread or direct extension from an underlying bony deposit.2–4,9 Extramedullary dissemination of myeloma, including to cutaneous sites, is usually associated with a very poor prognosis, with most patients dying within months of its occurrence.3,4,10
Histologic features The histologic features of primary cutaneous plasmacytoma and metastatic myeloma are similar. The infiltrate may be nodular or diffuse and is usually extensive, frequently involving the subcutaneous fat (Figs 29.232 and 29.233).4,10 A distinct grenz zone is commonly present. An interstitial pattern of growth has been described in cases of secondary cutaneous involvement. Although in some well-differentiated examples the plasma cell nature of the infiltrate is obvious, with characteristic clock-face nuclei, more often the tumor cells are atypical, having distinctly angulated and often molded cytoplasmic borders with pleomorphic nuclei containing conspicuous nucleoli and granular eosinophilic cytoplasm.3 Binucleate and multinucleate forms are often present, and mitotic figures may be abundant in less well-differentiated lesions.3 Intracytoplasmic inclusions (Russell bodies) and intranuclear inclusions (Dutcher bodies), although not usually prominent, may sometimes be present.5,10,19 Exceptionally, eosinophilic rhomboidal or needle-shaped crystals may be identified in the cytoplasm of tumor-associated histiocytes (crystal-storing histiocytosis), this being more often associated with plasma cell myeloma (Fig. 29.234).24–27 A case of secondary mucinosis has also been reported.28
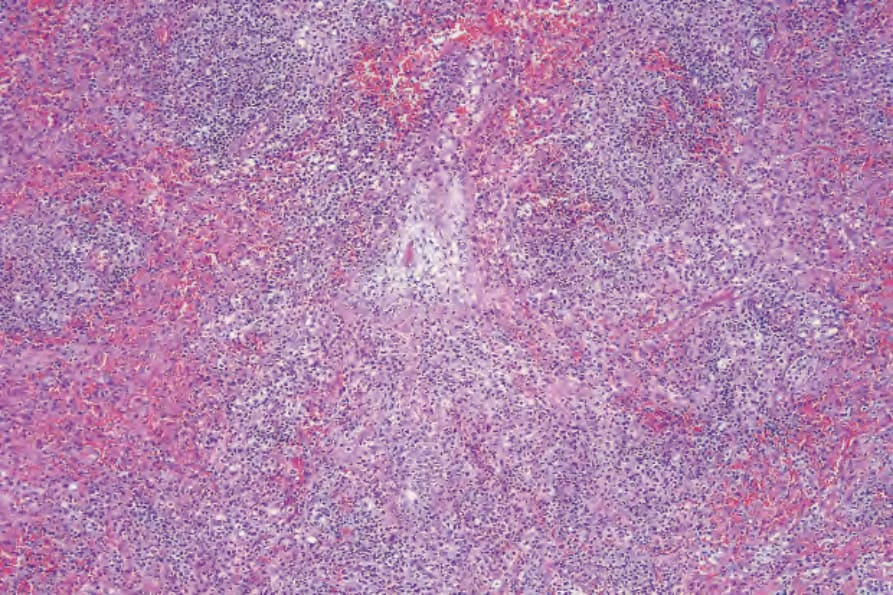
Fig. 29.228 Lymphomatoid granulomatosis: there is a multinodular tumor cell infiltrate.
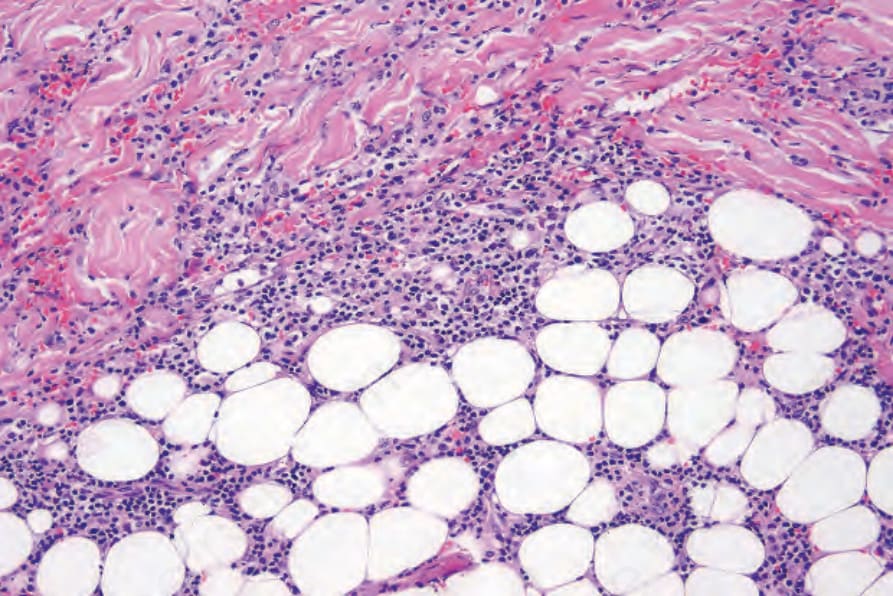
Fig. 29.229 Lymphomatoid granulomatosis: the infiltrate extends into the subcutaneous fat.
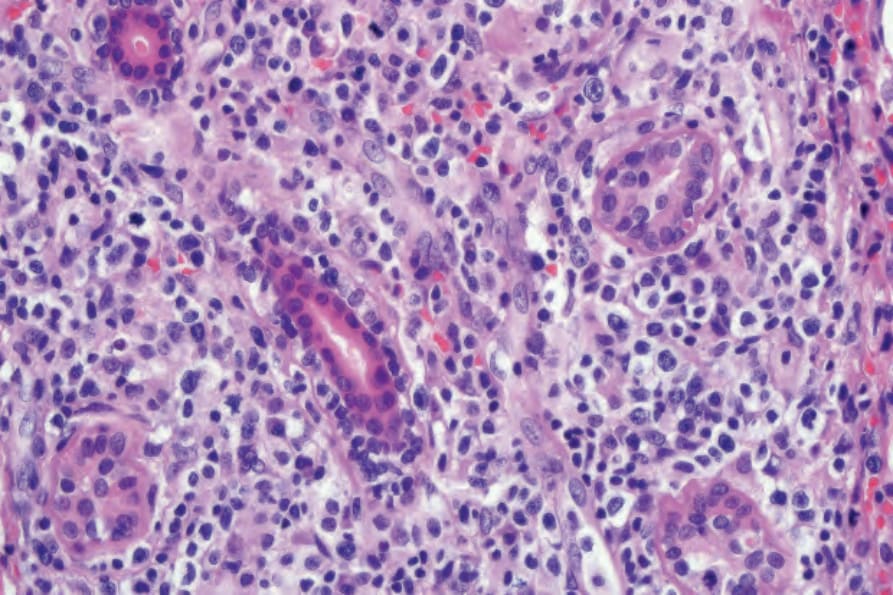
Fig. 29.230 Lymphomatoid granulomatosis: high-power view of dermis showing atypical lymphoid cells.
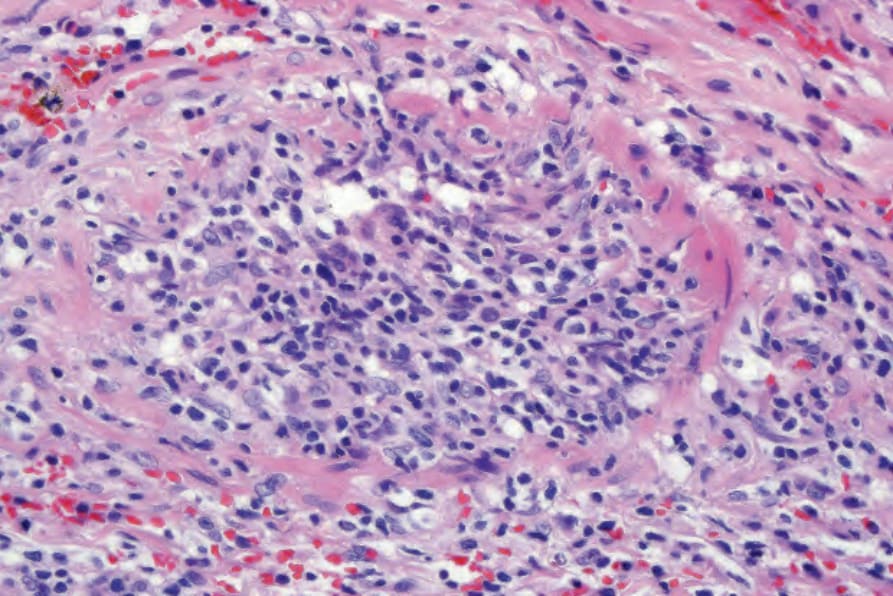
Fig. 29.231 Lymphomatoid granulomatosis: there is striking vascular involvement.
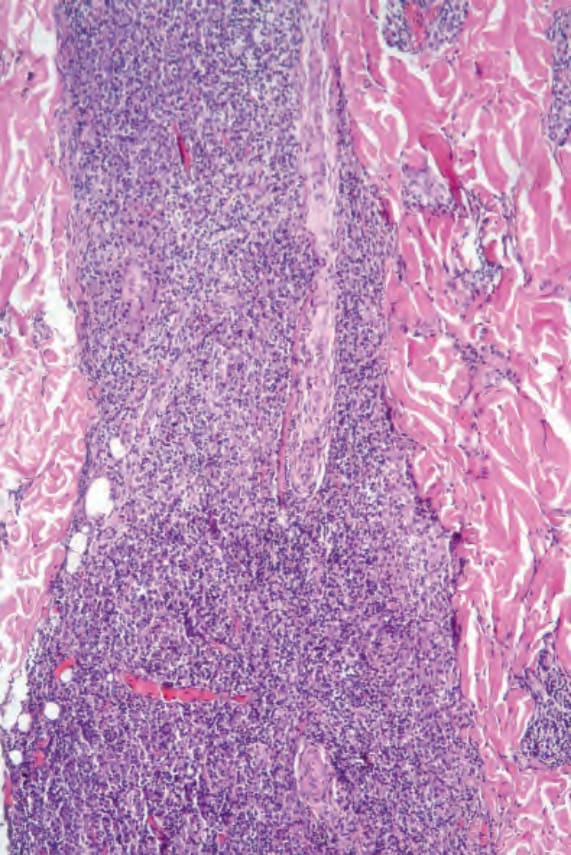
Fig. 29.232 Plasmacytoma: there is a dense dermal infiltrate. By courtesy of J. Cohen, MD, Dermatopathology Laboratory, Tucson, Arizona, USA.
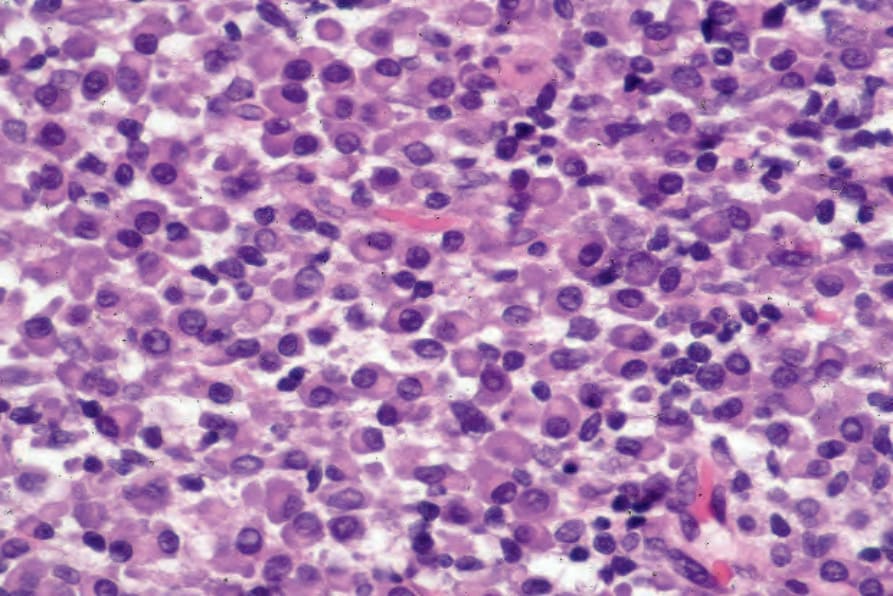
Fig. 29.233 Plasmacytoma: the infiltrate consists of an almost pure plasma cell population. By courtesy of J. Cohen, MD, Dermatopathology Laboratory, Tucson, Arizona, USA.
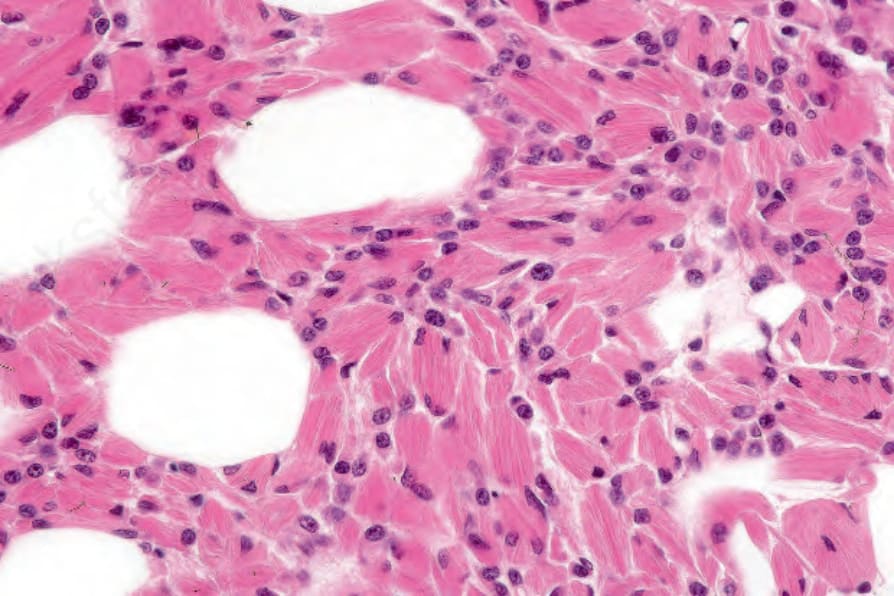
Fig. 29.234 Multiple myeloma: histiocytes containing needle-shaped crystals are evident – so-called crystal-storing histiocytosis. By courtesy of G. Pinkus, MD, Brigham and Women’s Hospital and Harvard Medical School, Boston, USA.
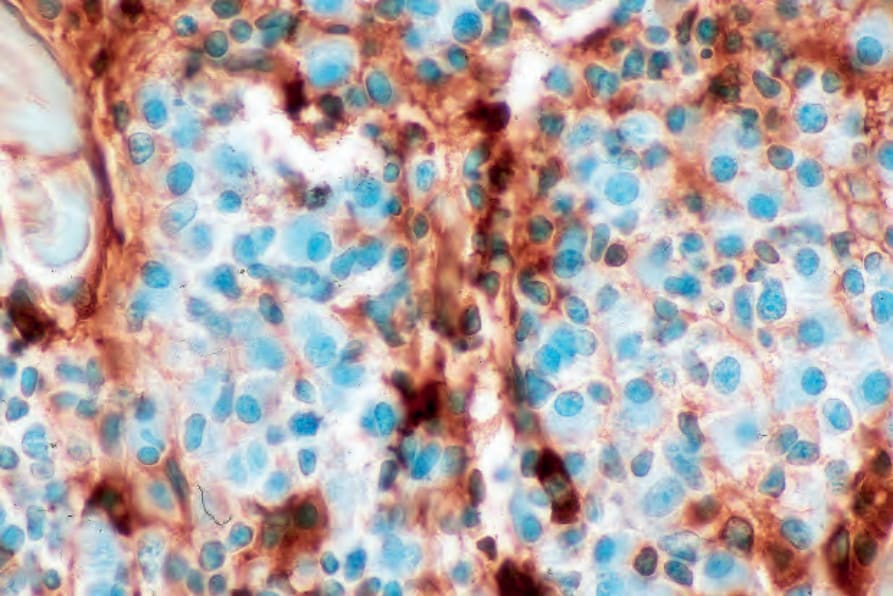
Fig. 29.237 Plasmacytoma: lambda light chain is absent. By courtesy of J. Cohen, MD, Dermatopathology Laboratory, Tucson, Arizona, USA.
Myelomatous deposits present as 1–5 cm in diameter, solitary, or more often multiple flesh-colored, red or violaceous nodules which affect the trunk, extremities, and face in decreasing order of frequency.1–3
Primary cutaneous plasmacytomas belong to the group of extraosseus (extramedullary) plasmacytomas. These are defined as neoplasms of plasma cells arising in tissues other than bone, and constitute 3–5% of all plasma cell neoplasms.11,12 Eighty percent of cases arise in the upper respiratory tract, but occurrence in the gastrointestinal tract, lymph nodes, bladder, brain, thyroid, testes, parotid, and skin is also documented.1 Consequently, primary cutaneous plasmacytomas are exceptional, with few genuine cases documented in the English-language literature.2,13 Lesions, which may be
By immunohistochemistry, the plasma cells express CD38, CD79a, and CD138 (Fig. 29.235). They are usually CD19 negative and often lack CD20 and common leukocyte antigen.4,13 Monotypic cytoplasmic Ig light chain is present (Figs 29.236 and 29.237). CD56 and cyclin D1 may be expressed, and occasionally there is positivity for EMA, HMB-45, and cytokeratin.2
1475 Intravascular large B-cell lymphoma
Differential diagnosis Differentiation of poorly differentiated forms of the disease from plasmablastic lymphoma is discussed in the section on plasmablastic lymphoma. Well-differentiated tumors are sometimes mistaken for plasma cell-rich conditions such as syphilis or Borrelia infection. If there is a significant background lymphocyte population, there may be confusion with primary cutaneous marginal zone B-cell lymphoma/immunocytoma or lymphoplasmacytic lymphoma. Distinction then rests on demonstrating a neoplastic component of lymphocytes, morphologically (marginal zone cells), with immunohistochemistry (CD20), or by flow cytometry. With poorly differentiated examples, the differential diagnosis can include high-grade lymphoma, carcinoma, or melanoma. In such instances, the immunohistochemistry panel of antibodies will require expansion.
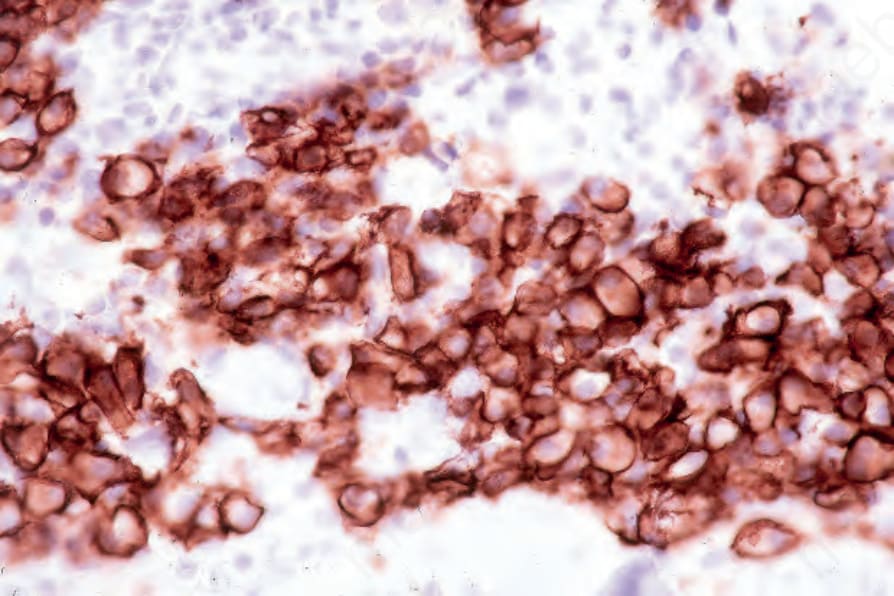
Fig. 29.235 Plasmacytoma: the tumor cells express CD138. By courtesy of J. Cohen, MD, Dermatopathology Laboratory, Tucson, Arizona, USA.
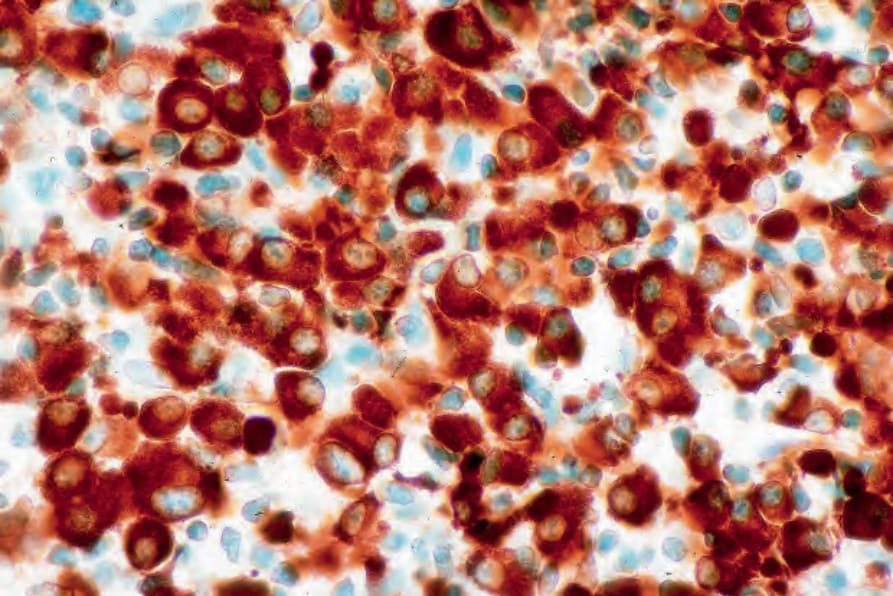
Fig. 29.236 Plasmacytoma: there is uniform kappa light chain expression. By courtesy of J. Cohen, MD, Dermatopathology Laboratory, Tucson, Arizona, USA.