Spitz 母斑的遺傳變異 (Genetic Alterations in Spitz Nevi)
- 如前所述,HRAS 突變僅見於少數 Spitz nevi。HRAS 突變型 Spitz nevi 常於染色體 11p 出現 HRAS 基因的增益 (gains),並與真皮內促結締組織增生 (desmoplastic) 及浸潤性 (infiltrative) 的生長模式相關。
- 約 50% 的 Spitz nevi 帶有染色體易位 (chromosomal translocations),產生含 ROS1、NTRK1、NTRK3、ALK、BRAF、RET 或 MET 之激酶結構域 (kinase domain) 的蛋白融合 (protein fusions)。這些激酶融合 (kinase fusions) 經由易位形成,使蛋白的 5′ 調控部分與 3′ 激酶結構域脫鉤 (decouple),從而導致不受限制的激酶活性。5′ 融合夥伴 (fusion partners) 多變。
- 當這些激酶融合事件出現於黑色素細胞腫瘤中時,會產生具 spitzoid 細胞學特徵的腫瘤,並隨特定激酶而具備一些額外可資鑑別的組織病理特徵。
- 帶有 ALK 融合的腫瘤通常為無黑色素性 (amelanotic),由飽滿、梭形 (plump, spindled) 的黑色素細胞構成,伴明顯的裂隙 (clefting) 及放射狀排列的紡錘形巢 (radially oriented, fusiform nests)(圖 25-255 與 25-256)。
- 在網狀真皮 (reticular dermis) 內常見垂直排列、交織的束狀結構 (vertically oriented, interweaving fascicles),周邊呈浸潤性生長模式,可藉 ALK 免疫染色凸顯(圖 25-257A 與 B)。
- 帶有 NTRK1 融合的腫瘤其束狀結構較短,且常沿毛囊 (hair follicles) 與小汗腺導管 (eccrine ducts) 延伸。NTRK3 腫瘤的黑色素細胞為上皮樣 (epithelioid) 或梭形 (spindled),於真皮內形成大型結節狀巢 (large nodular nests)。
生物學行為 (Biological Behavior)
- 被歸類為 MELTUMP 的病灶可分層為行為良好 (favorable) 與行為不良 (unfavorable) 兩類。
- 有絲分裂象 (mitoses) 的存在、其位於病灶基底附近的位置,以及病灶內的發炎細胞浸潤 (inflammatory cell infiltrate),皆為較常見於預後不良之 MELTUMP 的組織學特徵。
- 預後不良的 MELTUMP 被發現與顯著較高頻率的腫瘤相關死亡 (tumor-related death) 及/或淋巴結內大型轉移沉積物 (large metastatic deposits) 及/或內臟轉移 (visceral metastases) 相關。
- 相對地,行為良好的 MELTUMP 在追蹤超過 5 年後未觀察到轉移性疾病 (metastatic disease) 的證據。
BAP1 失活型 spitzoid 腫瘤 (BAP1-inactivated Spitzoid Tumors)
- 在帶有 BRAF 突變的母斑中,BRCA1 相關蛋白 1 (BRCA1-associated protein 1, BAP1) 腫瘤抑制基因 (tumor suppressor gene) 的突變或缺失,也可導致 spitzoid 腫瘤。
- 帶有種系 (germline) BAP1 突變的家族易罹患葡萄膜及皮膚黑色素瘤 (uveal and cutaneous melanoma)、腎細胞癌 (renal cell carcinoma)、間皮瘤 (mesothelioma),以及無黑色素性、spitzoid 腫瘤——後者由上皮樣至漿細胞樣 (epithelioid to plasmacytoid) 的黑色素細胞構成結節狀聚集,胞質呈淡嗜伊紅 (pale eosinophilic cytoplasm)。
- 這些 spitzoid 腫瘤通常位於真皮 (dermal),常具多核細胞 (multinucleated cells),且缺乏與 Spitz nevi 相關的典型表皮增生 (epidermal hyperplasia) 及 Kamino bodies。
- 由於它們通常源自常見後天性母斑 (common acquired nevi),故呈雙相 (biphasic) 表現:spitzoid 成分旁伴隨一群小型黑色素細胞(圖 25-258)。
- 可見強度不一的淋巴組織球性發炎細胞浸潤 (lymphohistiocytic inflammatory cell infiltrate)。有絲分裂活性通常偏低。
- 兩種黑色素細胞常皆帶有 BRAF 突變,而僅 spitzoid 成分帶有 BAP1 突變。
- 於傳統母斑 (conventional nevus) 區域進行 BAP1 免疫染色顯示正常的陽性核染色 (positive nuclear staining),而 spitzoid 成分則無核染色(圖 25-259A 與 B)。有時 spitzoid 成分可見明顯的胞質染色 (cytoplasmic staining),因為某些 BAP1 突變會破壞蛋白羧基末端 (carboxy-terminus) 的核定位序列 (nuclear localization sequence)。
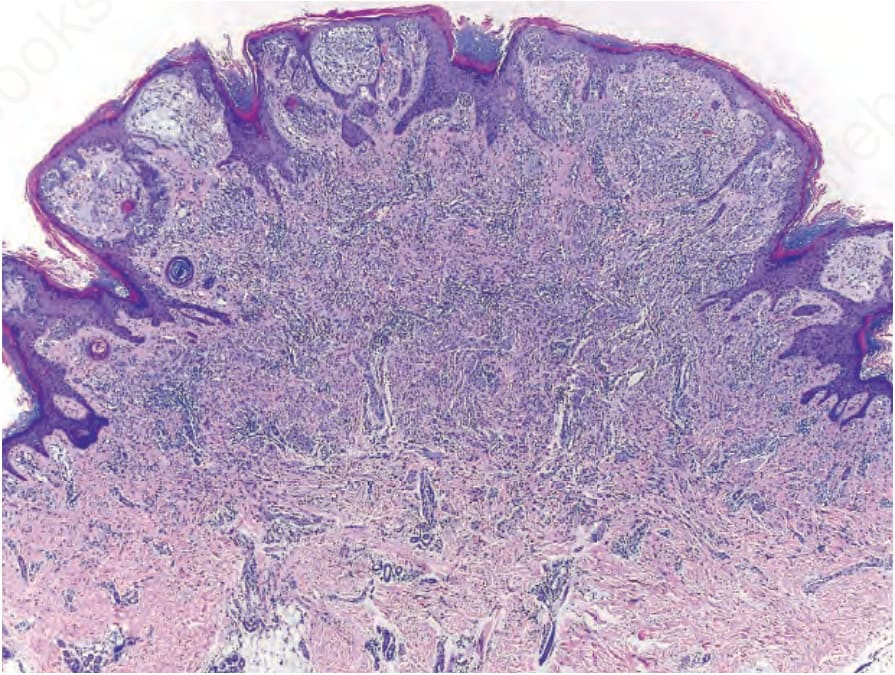
圖 25-255:帶有 ALK 融合的複合型 Spitz nevus (compound Spitz nevus):在不規則增生的表皮下方可見帶有裂隙的黑色素細胞巢 (nests of melanocytes with clefts)。黑色素細胞的巢與束狀結構自交界處流入網狀真皮,深部巢的尺寸逐漸縮小。
Fig. 25.255 Compound Spitz nevus with an ALK fusion: nests of melanocytes with clefts are present below an irregularly hyperplastic epidermis. Nests and fascicles of melanocytes stream from the junction into the reticular dermis with diminution of nest size in the deep portion.
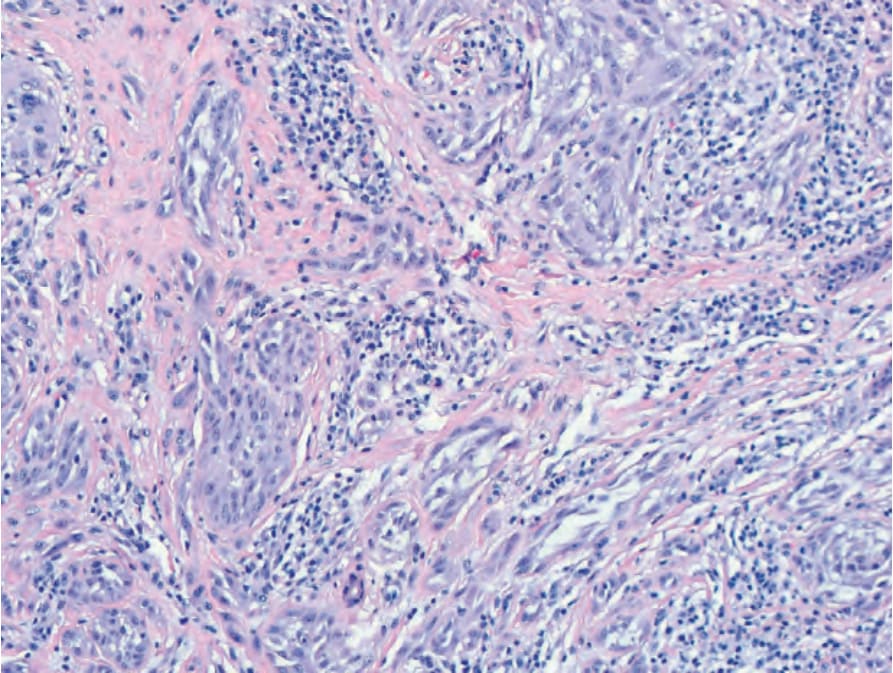
圖 25-256:帶有 ALK 融合的 Spitz nevus:網狀真皮內由上皮樣與梭形黑色素細胞構成的束狀結構,呈垂直排列並具明顯裂隙 (prominent clefting)。
Fig. 25.256 Spitz nevus with an ALK fusion: fascicles of epithelioid and spindled melanocytes with prominent clefting in vertical orientation in the reticular dermis.
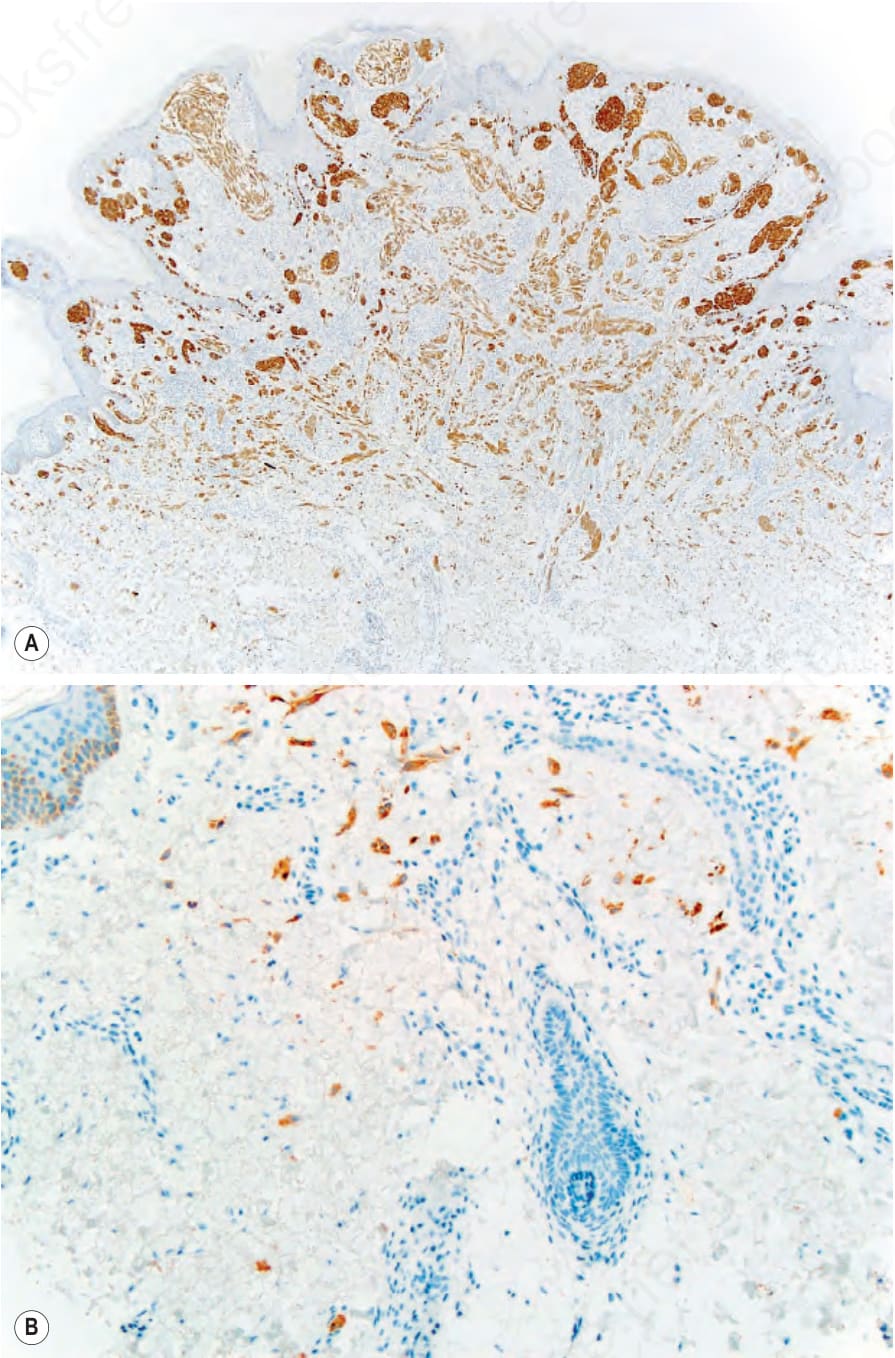
圖 25-257:(A) 帶有 ALK 融合的 Spitz nevus:ALK 免疫染色凸顯整個腫瘤,包括周邊的浸潤性模式 (infiltrative pattern),可見單一陽性黑色素細胞延伸至遠離腫瘤主體之處 (B 或插圖)。
Fig. 25.257 (A) Spitz nevus with an ALK fusion: ALK immunostaining highlights the entire neoplasm, including an infiltrative pattern at the periphery, with single positive melanocytes extending far from the main portion of the neoplasm (B or inset).
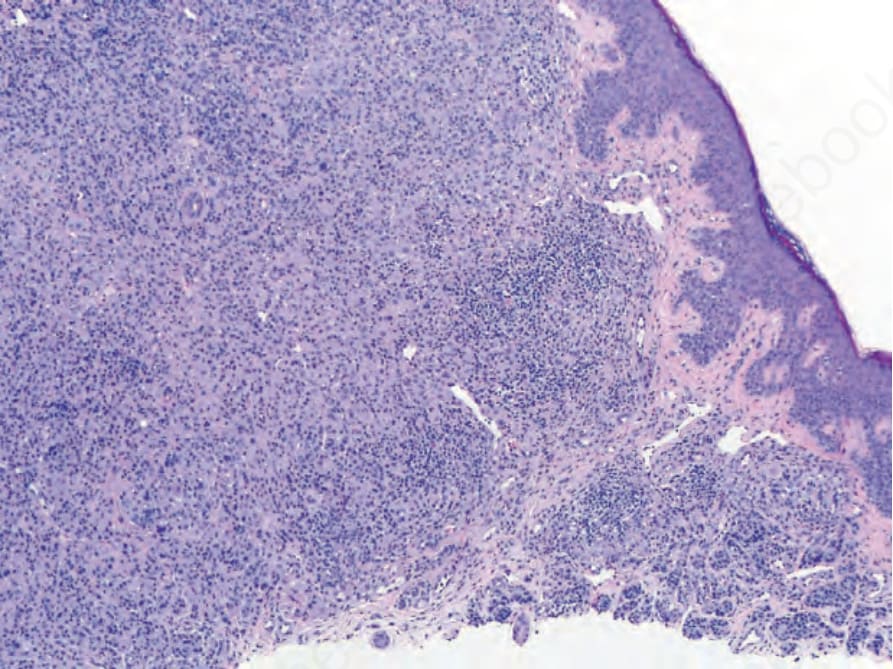
圖 25-258:BAP1 失活型 spitzoid 腫瘤 (BAP1-inactivated spitzoid tumor):在 spitzoid 細胞的大型結節狀聚集旁,可見小型圓形黑色素細胞的巢,代表保有功能性 BAP1 蛋白 (positive nuclear BAP1 staining) 的前驅母斑 (precursor nevus) 殘餘細胞群。
Fig. 25.258 BAP1-inactivated spitzoid tumor: adjacent to the large nodular collection of spitzoid cells, there are nests of small, round melanocytes representing a remnant population of the precursor nevus that retains a functional BAP1 protein (positive nuclear BAP1 staining).
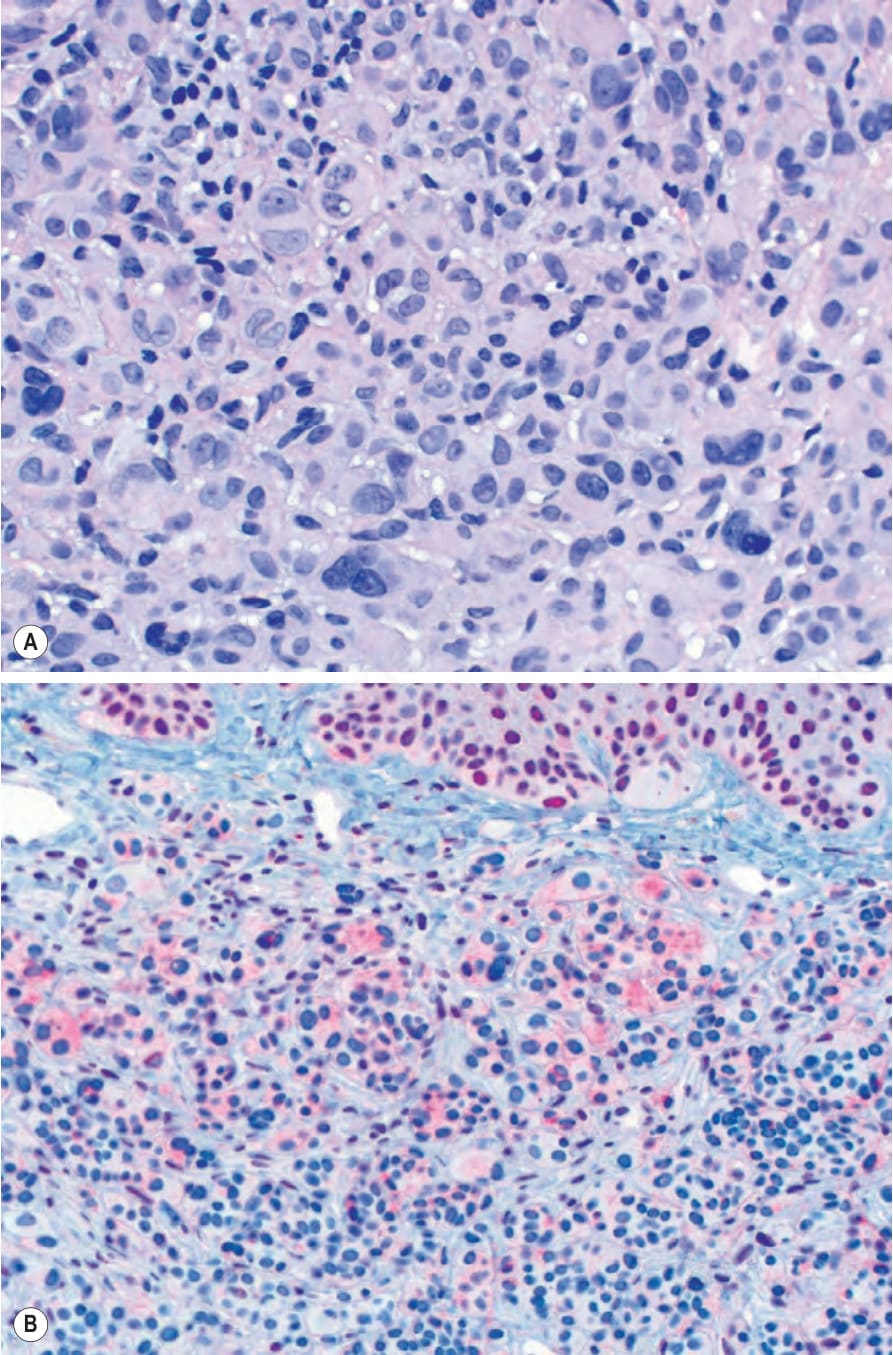
圖 25-259:(A) BAP1 失活型 spitzoid 腫瘤:大型上皮樣黑色素細胞具豐富淡染胞質 (abundant pale cytoplasm),於真皮內形成結節。可見眾多多核黑色素細胞。(B, 插圖) BAP1 染色顯示上覆表皮角質細胞 (epidermal keratinocytes) 核染色完整,而上皮樣黑色素細胞核染色缺失。
Fig. 25.259 (A) BAP1-inactivated spitzoid tumor: large epithelioid melanocytes with abundant pale cytoplasm form nodules in the dermis. Numerous multinucleated melanocytes are present. (B, inset) BAP1 stain showing intact nuclear staining of overlying epidermal keratinocytes and loss of nuclear staining in epithelioid melanocytes.
黑色素細胞母斑的分子病理 (The Molecular Pathology of Melanocytic Nevi)
| 基因 | 變異 (Aberration) | 細胞路徑 (Cellular Pathway) | 母斑類型 (Nevus type(s)) | 組織病理特徵 |
|---|---|---|---|---|
| BRAF | 點突變 (Point mutation) | MAP kinase | 後天性母斑 (Acquired nevi) | -巢狀模式 (Nested pattern) |
| BRAF | 重排 (Rearrangement) | MAP kinase | 先天性母斑 (Congenital nevi)、Spitz nevi | |
| NRAS | 點突變 (Point mutation) | MAP kinase、PI3 kinase | 先天性母斑 (Congenital nevi) | |
| HRAS | 點突變、拷貝數增益 (Point mutation, copy number gain) | MAP kinase、PI3 kinase | Spitz nevi | -真皮內、促結締組織增生、浸潤性結構 (Intradermal, desmoplastic, infiltrative architecture) |
| BAP1 | 截短型非錯義及錯義突變、缺失 (Truncating non- and missense mutation, deletion) | BRCA1;染色質重塑 (chromatin remodeling) | BAP1 失活型 spitzoid 母斑 (BAP1-inactivated spitzoid nevi) | -上皮樣、漿細胞樣黑色素細胞 (Epithelioid, plasmacytoid melanocytes) -無黑色素性、真皮內腫瘤 (Amelanotic, intradermal tumor) -多核細胞 (Multinucleated cells) |
| GNAQ/GNA11 | 點突變 (Point mutation) | PKC、MAP kinase | 藍母斑 (Blue nevi)、太田母斑與伊藤母斑 (Nevi of Ota and Ito)、黑色素細胞瘤 (melanocytomas)、葡萄膜母斑 (uveal nevi) | -高色素性、樹突狀真皮黑色素細胞 (Hyperpigmented, dendritic dermal melanocytes) |
| ALK | 重排 (Rearrangement) | MAP kinase、PI3 kinase、STAT3 | Spitz nevi | -具明顯裂隙的紡錘形黑色素細胞 (Fusiform melanocytes with prominent clefting) -相互連接的束狀巢狀模式 (Interconnecting fascicular nesting pattern) |
| NTRK1 | 重排 (Rearrangement) | MAP kinase、PI3 kinase、PLCγ1、STAT3 | Spitz nevi | -具短束狀結構的紡錘形巢 (Fusiform nests with short fascicles) -附屬器延伸 (Adnexal extension) |
| NTRK3 | 重排 (Rearrangement) | MAP kinase、PI3 kinase、STAT3 | Spitz nevi | -上皮樣或紡錘形 (Epithelioid or fusiform) -大型真皮結節/巢 (Large dermal nodules/nests) |
| MET | 重排 (Rearrangement) | MAP kinase、PI3 kinase、STAT3 | Spitz nevi | |
| RET | 重排 (Rearrangement) | MAP kinase、PI3 kinase、PLCγ1、STAT3 | Spitz nevi | |
| ROS1 | 重排 (Rearrangement) | MAP kinase、PI3 kinase、STAT3 | Spitz nevi |
- 剩餘的野生型 (wild type) BAP1 等位基因的喪失,通常經由 BAP1 所在的染色體 3p 部分缺失、整條第 3 號染色體喪失,或拷貝數中性的雜合性喪失 (copy number neutral loss of heterozygosity) 而發生。
- 這些同時帶有 BAP1 與 BRAF 突變的腫瘤代表一種中間型腫瘤 (intermediate neoplasms),即在一個 BRAF 突變母斑內,因 BAP1 的雙等位基因失活 (bi-allelic inactivation) 而發生了第二次克隆性擴增 (clonal expansion)。初步資料顯示,在此進展階段它們的轉移潛能 (metastatic potential) 極低。
- 帶有種系 BAP1 突變的病人通常具有多個此類病灶。具有這些組織病理特徵的散發性腫瘤 (sporadic tumors) 則帶有兩個 BAP1 等位基因的體細胞失活 (somatic inactivation),且與 BAP1 的種系突變無關。
註解 (Comment)
- 使用 MELTUMP 一詞意味著病理醫師存在診斷上的不確定性。如同 ‘atypical Spitz nevus’,它必然由良性與惡性病灶的混合所構成。
- 極有可能其中絕大多數此類病灶,可藉由檢查額外切片 (extra sections)、更深層次 (deeper levels)、適當使用免疫組織化學 (immunohistochemistry)、分子遺傳學技術 (molecular genetic techniques),以及徵詢該領域專家意見而獲得更恰當的分類——除非僅收到部分切片標本 (partial biopsy specimen)。
- 雖然應盡量避免使用此一診斷類別……
致癌驅動因子 (Oncogenic Drivers)
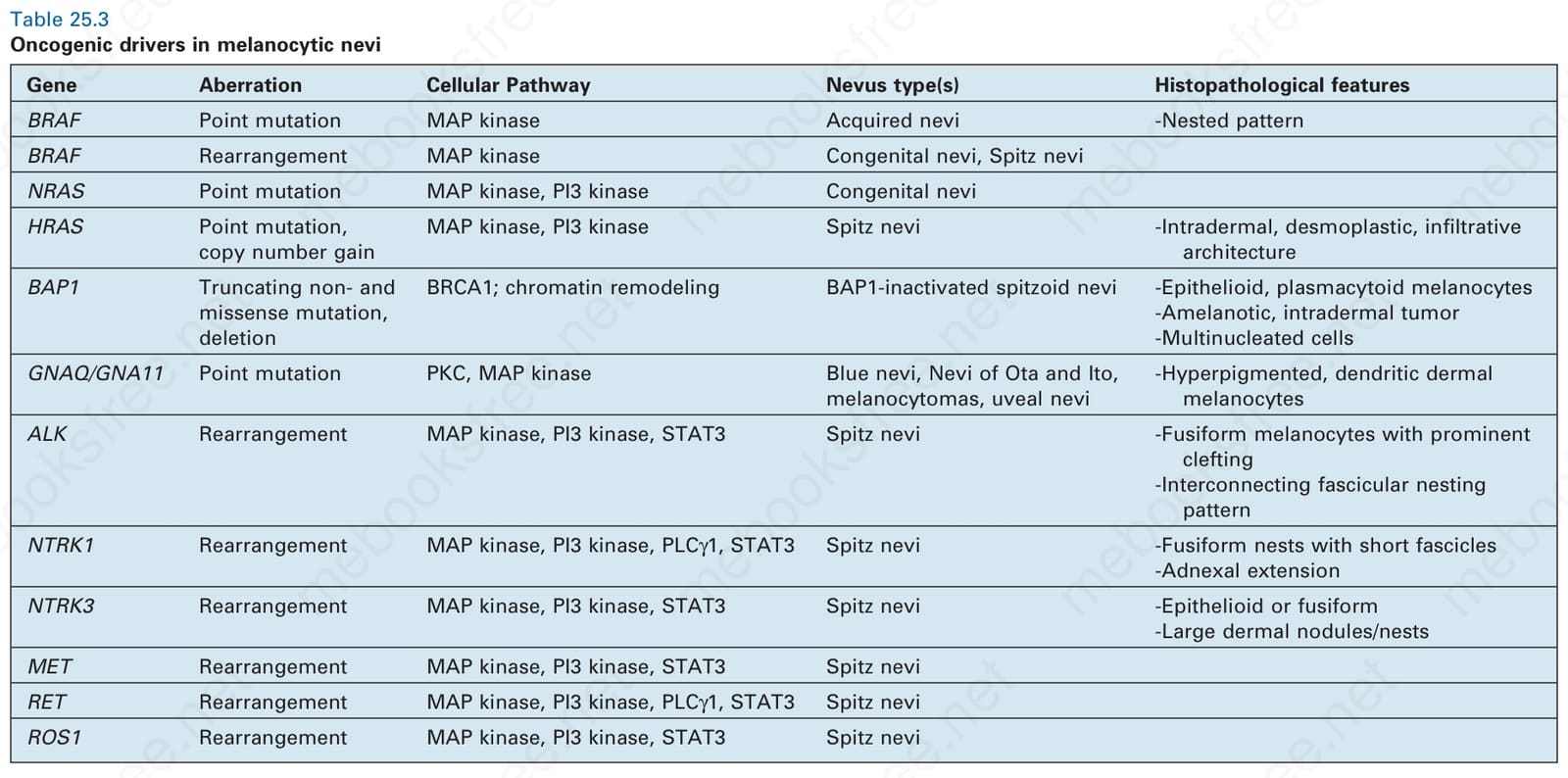
表 25-3:黑色素細胞母斑中的致癌驅動因子 (oncogenic drivers in melanocytic nevi)。
Table 25.3 Oncogenic drivers in melanocytic nevi
致癌突變與細胞衰老 (Oncogenic Mutations and Senescence)
- 黑色素瘤 (melanoma) 與母斑中存在相同的致癌突變 (oncogenic mutations),顯示這些突變於黑色素細胞惡性轉化 (malignant transformation) 的早期即已出現。雖然它們能誘導細胞短暫增殖 (transient proliferation),但不足以造成完全轉化,後者尚需額外協同的遺傳變異。
- 此一觀念獲得體外實驗 (in vitro experiments) 支持:導入活化的致癌基因(如 RAS 家族成員或下游激酶 BRAF)本身不足以轉化正常人類細胞。
- 強健的細胞防護機制 (cellular safeguard mechanisms) 存在,以限制那些因反覆暴露於紫外線輻射 (UV radiation)、外源性致突變物 (exogenous mutagens) 與活性氧物種 (reactive oxygen species) 而獲得突變的細胞增殖。
致癌基因誘導的衰老 (Oncogene-induced Senescence)
- 例如,信號組分突變所造成的 MAP-激酶路徑 (MAP-kinase pathway) 等信號路徑的組成性活化 (constitutive activation),會導致異常偏高的組成性信號通量 (constitutive signaling flux),此與生長因子暴露所誘導的信號模式不同。
- 在生理條件下,生長信號透過複雜的回饋機制 (feedback mechanisms) 迅速衰減,例如藉由受體內化 (receptor internalization) 及關鍵信號組分之抑制因子的上調來抑制輸入信號(圖 25-260A)。
- 相對地,致癌突變所造成的路徑過度活化,被認為會觸發類似(或許還有額外的)回饋機制,以對抗促生長效應(圖 25-260B)。這涉及細胞週期抑制因子 (cell cycle inhibitors) 如 p16 與 p21,以及其他「致癌壓力」(oncogenic stress) 感測器如 p14/ARF 的誘導。
- p16 與 p21 是週期蛋白依賴性激酶 (cyclin-dependent kinases) 的抑制因子,會使 RB 腫瘤抑制蛋白失活;而 p14/ARF 作用於 p53 上游,阻斷其降解。致癌基因活化會啟動 RB 與 p53 路徑,從而抑制增殖。
- 上述細胞週期抑制蛋白的表現可在大多數母斑中偵測到。然而值得注意的是,例如母斑內 p16 的表現模式可能呈斑塊狀 (patchy),部分腫瘤性黑色素細胞表現強烈,其他則否。這可能顯示病灶內並非所有細胞都依賴相同的因子或機制來停止增殖。
- 此外,某些細胞週期抑制因子也被發現於黑色素瘤中有不同程度的表現,顯示這些細胞可能已對其效應產生抗性。
- CDKN2A 基因編碼 p16 與 p14,在黑色素細胞腫瘤形成 (melanocytic neoplasia) 中似乎扮演特別重要的角色。CDKN2A 所在的染色體 9p21 是黑色素瘤中最常見的染色體喪失區域,近期一項研究顯示,CDKN2A 的純合性喪失 (homozygous loss) 常發生於母斑轉變為侵襲性黑色素瘤 (invasive melanoma) 的過程中。
- 根據此「致癌基因誘導衰老」(oncogene-induced senescence) 的概念,獲得致癌突變的細胞之生長停滯 (growth arrest) 表現為一相對急性的現象。然而,母斑中所見的細胞週期停滯似乎具有相當的延遲性 (latency)。為了臨床上可被偵測,母斑必須由數千個細胞組成,這些細胞由一個前驅細胞 (progenitor cell) 經過相當多次細胞分裂產生。
- 諸如 BRAF 等基因的致癌突變存在於大多數母斑細胞中,顯示這些細胞必定在突變存在下增殖,並將其在族群中傳播。上述依賴「立即偵測致癌基因活化並以跳脫的保險絲 (tripped fuse) 形式觸發生長停滯」的致癌基因誘導衰老機制,無法解釋此一延遲性。
- 有可能此種立即型衰老 (immediate-type senescence) 確實存在於體內,但帶有致癌突變的細胞會立即或僅經數輪分裂後即停滯,因而後續病灶在臨床上無法偵測。這引出一個耐人尋味的可能性:在易發生多發性母斑的病人中,致癌基因誘導衰老可能並未完全發揮功能。
- 若其中一道屏障失效,似乎會出現額外的衰老機制。例如,CDKN2A 基因功能喪失突變 (loss-of-function mutations) 純合的病人仍會發生母斑,雖然這些母斑通常體積較大。在此類母斑中,p16/p14 路徑以外的不同機制負責生長停滯。
複製性/端粒誘導的衰老 (Replicative or Telomere-induced Senescence)
- 母斑生長停滯的另一替代機制為複製性或端粒誘導的衰老 (replicative or telomere-induced senescence)(圖 25-260C)。端粒 (Telomeres) 是位於染色體末端的複雜結構,藉由形成套索結構 (lariat structure) 以串聯 DNA 重複序列、並由核蛋白基質 (matrix of nucleoproteins) 保護,來隔離 DNA 的開放末端。
- 在大多數體細胞中,端粒隨每次細胞分裂而縮短,於 60–70 次分裂後達到一臨界長度 (critical length),使 DNA 開放末端暴露,並觸發一個使細胞永久停滯的 DNA 損傷信號。
- 若此關鍵信號級聯 (signal cascade) 受損,細胞可繼續複製,並在姊妹染色單體 (sister chromatids) 之間形成開放端粒的端對端融合 (end-to-end fusions),產生雙著絲粒染色體 (bicentric chromosomes)。
- 這會導致染色體斷裂/融合/橋接循環 (breakage/fusion/bridge cycles),造成染色體增益與缺失。隨之而來的基因組混亂 (genomic chaos) 導致高頻率的細胞死亡,稱為危機 (crisis)。
- 危機一方面對於那些因遭受過多遺傳變異而無法存活、基因上已不適格的細胞,作為一種腫瘤預防機制 (tumor preventative mechanism);另一方面,它產生新的遺傳變異株,其中部分可能獲得增強的增殖能力。
- 轉化克隆 (transformed clones) 的出現源自罕見細胞,這些細胞藉由獲得重新穩定其端粒的能力(例如透過端粒酶 (telomerase) 的活化,此酶以其自身的 RNA 模板延長端粒)而成功逃脫危機。
- TERT promoter 的突變可促進端粒酶的產生以克服端粒誘導的衰老,並見於包括黑色素瘤在內的眾多癌症中。近期研究顯示,TERT promoter 突變在尚未完全惡性轉化為黑色素瘤之前,即可於包括異型增生母斑 (dysplastic nevi) 在內的中間階段黑色素細胞腫瘤中偵測到。
- TERT 活化在黑色素細胞腫瘤演化中如此早期出現,顯示母斑細胞可能持續分裂直到端粒耗盡為止。
- 大多數母斑由相對少量的細胞構成,估計介於 10⁵ 至 10⁶ 個細胞之間,遠低於 60 次細胞分裂之複製極限 (replicative limit) 所預期的數目。此預期與觀察到的細胞數目之間的顯著差異,顯示存在非常顯著的損耗因子 (attritional factors),必定消除了母斑中許多細胞。免疫系統消除細胞及/或細胞自主機制 (cell-autonomous mechanisms) 如細胞凋亡 (apoptosis),是可能的候選機制。
- 此種母斑形成與維持的動態情境(相對於穩定的衰老細胞族群),可解釋母斑外觀在縱貫研究 (longitudinal studies) 中的變化,也能解釋為何母斑往往在 30 歲之後消退。一旦細胞達到其複製壽命 (replicative life span),增殖與損耗之間的平衡將傾向損耗。只有那些藉由 TERT promoter 突變克服此屏障的病灶,才能繼續增殖(並獲得額外突變以朝黑色素瘤演化)。
- 複製壽命與母斑表型 (nevus phenotype) 相關。出生後獲得的母斑通常不超過 2 cm,而當端粒較長時於子宮內 (in utero) 發育的母斑則可達到顯著更大的尺寸。這顯示起始黑色素細胞 (initiating melanocyte) 中的端粒大小會影響後續母斑的最大尺寸,且有關母斑尺寸與密度的研究已發現其與端粒長度 (telomere length) 的關聯。
- 在母斑及結腸腺瘤 (colon adenomas) 中已顯示出額外的衰老機制,其依賴一種涉及白介素 6 及 8 (interleukins 6 and 8) 的致癌基因誘導發炎反應 (oncogene-induced inflammatory response)。
- 衰老與發炎之間的關聯頗為有趣,因為許多母斑——尤其是異型增生母斑 (dysplastic nevi)——顯示慢性發炎浸潤 (chronic inflammatory infiltrate)。
DNA 損傷誘導的衰老 (DNA Damage-induced Senescence)
- DNA 損傷 (DNA damage) 也可觸發生長停滯。諸如 RAS 家族成員等致癌基因的突變會導致 DNA 複製錯誤增加,造成 DNA 損傷。
- 複製起點 (replication origins) 的錯誤激發會產生異常 DNA 結構,觸發關鍵檢查點 (checkpoints),通常以 p53 依賴的方式使細胞停滯,並在問題無法經 DNA 修復機制解決時永久停止增殖(見圖 25-260)。若問題無法修復,細胞即進入永久性生長停滯。
- 此類衰老被稱為 DNA 損傷誘導的衰老 (DNA damaged-induced senescence)。此機制已在數種癌前病灶 (precancerous lesions) 中得到證實,包括黑色素細胞母斑。
- DNA 損傷誘導衰老的情境意味著,母斑可帶有具超越單純點突變之遺傳變異的細胞,這些變異源自複製錯誤或染色體錯誤分離 (missegregation)。然而,在這些細胞中誘導衰老限制了它們的克隆性擴增(圖 25-261A)。
- 相對地,在黑色素瘤中這些檢查點喪失,允許具異常基因組的黑色素細胞增殖,並從中選擇出具有利基因組組成的克隆(圖 25-261B)。此情境可解釋為何克隆性染色體畸變 (clonal chromosomal aberrations) 在母斑中大多缺如,卻在黑色素瘤中非常常見(圖 25-262)。
- 總結而言,目前的證據強烈顯示,數種獨立機制限制了那些由強效致癌基因之活化突變所啟動的黑色素細胞之增殖,並協同提供一道對抗癌症形成的強健屏障。即使一道機制失效,後備機制仍可防止惡性轉化。此概念可解釋為何帶有 CDKN2A 功能喪失等位基因 (loss-of-function alleles) 的個體仍會發生母斑(儘管尺寸較大)。然而,若其中一道衰老機制受損,可能隨之而來黑色素瘤風險增加,這解釋了為何遺傳性 CDKN2A 突變是黑色素瘤的強烈風險因子。

圖 25-260:衰老機制的示例。(A) 在生理情況下,生長信號沿 MAP-激酶路徑傳遞,在無抑制信號時導致細胞分裂。(B) 若該路徑被過度活化(例如於 RAS 層次的突變),衰老媒介因子如 p16 即被誘導並抑制細胞週期進入。(C) 當一群細胞持續分裂、因端粒侵蝕 (telomere erosion) 耗盡其複製潛能時,便發生複製性衰老 (replicative senescence)。(D) DNA 損傷誘導的衰老由細胞週期中的隨機遺傳變異所致,伴隨後續的永久性生長停滯或細胞凋亡。
Fig. 25.260 Examples of mechanisms of senescence. (A) Under physiological circumstances, a growth signal is relayed down the MAP-kinase pathway and in the absence of inhibitory signals results in cell division. (B) If the pathway is hyperactivated, for example, by a mutation at the level of RAS, mediators of senescence such as p16 become induced and inhibit cell cycle entry. (C) Replicative senescence ensues when a population of cells continues to divide and exhausts its replicative potential due to telomere erosion. (D) DNA damaged-induced senescence is caused by random genetic alterations during the cell cycle with subsequent permanent growth arrest or apoptosis.
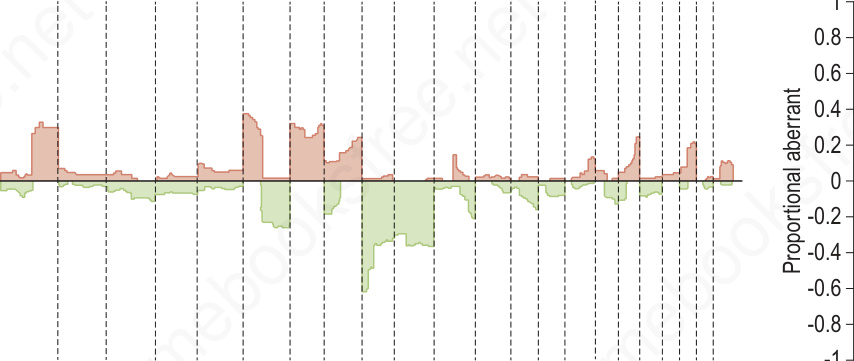
圖 25-262:黑色素瘤(惡性)與母斑(良性)的比較基因組雜交 (comparative genomic hybridization):左側黑色素瘤 (n = 133) 伴隨多處聚集的拷貝數增益與喪失。相對地,右側母斑 (n = 54) 顯示極微小的變化,唯一例外為 Spitz nevi 中包含 HRAS 基因的 11p 拷貝數增加或擴增 (amplification)。在具挑戰性的病例中,可利用這些差異以多重 FISH 檢測 (multiplexed FISH assays) 來支持黑色素瘤或母斑的診斷。
Fig. 25.262 Comparative genomic hybridization of melanoma (malignant) and nevi (benign): on the left, melanomas (n = 133) are associated with multiple copy number gains and losses that cluster. In contrast, the nevi (n = 54) on the right show minimal changes with the exception of the 11p copy number increase or amplification in Spitz nevi which includes the HRAS gene. These differences can be exploited using multiplexed FISH assays to support the diagnosis of melanoma or nevus in challenging cases.