Genetic alterations in Spitz nevi
Genetic alterations in Spitz nevi
As noted above, HRAS mutations occur in a minority of Spitz nevi. HRAS mutant Spitz nevi frequently have gains of the HRAS gene at chromosome 11p and are associated with an intradermal desmoplastic and infiltrative growth pattern.5 Approximately 50% of Spitz nevi harbor chromosomal translocations resulting in protein fusions containing the kinase domain of ROS1, NTRK1, NTRK3, ALK, BRAF, RET, or MET.6–8 These kinase fusions form through translocations which decouple the 5′ regulatory portion of the proteins from the 3′ kinase domain, thereby leading to unrestrained kinase activity. The 5′ fusion partners are variable. When present in melanocytic tumors, these kinase fusion events produce tumors with spitzoid cytological features, with some additional distinguishing histopathological features associated with particular kinases. Tumors with ALK fusions are usually amelanotic and have plump, spindled melanocytes with prominent clefting and radially oriented, fusiform nests (Figs 25.255 and 25.256).9 Vertically oriented, interweaving fascicles are often present in the reticular dermis, with an infiltrative growth pattern at the periphery that can be highlighted by ALK immunostaining (Figs 25.257A & B). Tumors with NTRK1 fusions have shorter fascicles and frequent extension along hair follicles and eccrine ducts. NTRK3 tumors have epithelioid or spindled melanocytes that form large nodular nests in the dermis.8
Biological behavior Lesions designated as MELTUMP have been stratified as those with favorable and those with unfavorable behavior.1 The presence of mitoses, their location near the base of the lesion, and inflammatory cell infiltrate within the lesion are histologic features that are more frequently found in MELTUMP with unfavorable prognosis. MELTUMP with unfavorable outcome were found to be associated with significantly higher frequency of tumor-related death and/or large metastatic deposits in the lymph nodes and/or visceral metastases.1 In contrast, no evidence of metastatic disease after more than 5 years of follow-up was observed in MELTUMP with favorable behavior.1
Mutation or loss of the BRCA1-associated protein 1 (BAP1) tumor suppressor gene in nevi with BRAF mutations can result in spitzoid tumors as well. Families with germline BAP1 mutations are predisposed to uveal and cutaneous melanoma, renal cell carcinoma, mesothelioma, and amelanotic, spitzoid tumors with nodular collections of epithelioid to plasmacytoid melanocytes with pale eosinophilic cytoplasm.10 These spitzoid tumors are typically dermal, often have multinucleated cells, and lack the typical epidermal hyperplasia and Kamino bodies associated with Spitz nevi. As they typically arise from common acquired nevi, they present in biphasic fashion with a component of small melanocytes adjacent to the spitzoid component (Fig. 25.258). A lymphohistiocytic inflammatory cell infiltrate of variable intensity may be seen. Mitotic activity is usually low. Both types of melanocytes frequently harbor BRAF mutations, while the spitzoid component alone has a BAP1 mutation. Immunostaining for BAP1 in areas of conventional nevus shows normal positive nuclear staining, while the spitzoid component exhibits no nuclear staining (Fig. 25.259A & B). Sometimes cytoplasmic staining is evident in the spitzoid component because some BAP1 mutations
Comment Use of the term MELTUMP implies diagnostic uncertainty on behalf of the pathologist. As with ‘atypical Spitz nevus’, it must be composed of an admixture of benign and malignant lesions. It is highly likely that the overwhelming majority of such lesions can be more appropriately classified by the examination of extra sections, deeper levels, the appropriate use of immunohistochemistry, molecular genetic techniques, and the opinion(s) of expert(s) in the field sought unless only a partial biopsy specimen has been received. Although one should try and avoid using this diagnostic category
1305 The molecular pathology of melanocytic nevi
Gene Aberration Cellular Pathway Nevus type(s) Histopathological features
BRAF Point mutation MAP kinase Acquired nevi -Nested pattern
BRAF Rearrangement MAP kinase Congenital nevi, Spitz nevi
NRAS Point mutation MAP kinase, PI3 kinase Congenital nevi
HRAS Point mutation, copy number gain
MAP kinase, PI3 kinase Spitz nevi -Intradermal, desmoplastic, infiltrative
architecture
BAP1 Truncating non- and missense mutation, deletion
BRCA1; chromatin remodeling BAP1-inactivated spitzoid nevi -Epithelioid, plasmacytoid melanocytes -Amelanotic, intradermal tumor -Multinucleated cells
GNAQ/GNA11 Point mutation PKC, MAP kinase Blue nevi, Nevi of Ota and Ito, melanocytomas, uveal nevi
-Hyperpigmented, dendritic dermal
melanocytes
ALK Rearrangement MAP kinase, PI3 kinase, STAT3 Spitz nevi -Fusiform melanocytes with prominent
clefting -Interconnecting fascicular nesting
pattern
NTRK1 Rearrangement MAP kinase, PI3 kinase, PLCγ1, STAT3 Spitz nevi -Fusiform nests with short fascicles -Adnexal extension
NTRK3 Rearrangement MAP kinase, PI3 kinase, STAT3 Spitz nevi -Epithelioid or fusiform -Large dermal nodules/nests
MET Rearrangement MAP kinase, PI3 kinase, STAT3 Spitz nevi
RET Rearrangement MAP kinase, PI3 kinase, PLCγ1, STAT3 Spitz nevi
ROS1 Rearrangement MAP kinase, PI3 kinase, STAT3 Spitz nevi
corrupt the nuclear localization sequence at the carboxy-terminus of the protein.10 Loss of the remaining wild type BAP1 allele typically occurs by deletion of the portion of chromosome 3p where BAP1 resides, loss of the entire chromosome 3, or by copy number neutral loss of heterozygosity. These tumors with both BAP1 and BRAF mutations represent intermediate neoplasms in which a second clonal expansion has occurred within a BRAF mutant nevus as a consequence of bi-allelic inactivation of BAP1.10 Preliminary data suggest they have minimal metastatic potential at this stage of progression.11 Patients with germline BAP1 mutations typically have multiple of these lesions. Sporadic tumors with these histopathological features
have somatic inactivation of both BAP1 alleles, and are not associated with germline mutations of BAP1.
The presence of identical oncogenic mutations in melanoma and nevi indicates that these mutations arise early during the malignant transformation of melanocytes. While they can induce a transient proliferation of cells, they are not sufficient for full transformation, which requires additional cooperating genetic alterations. This notion is supported by in vitro experiments in which the introduction of activated oncogenes such as RAS family members or the downstream kinase BRAF are insufficient by themselves to transform normal human cells.12,13 Robust cellular safeguard mechanisms exist to restrict the proliferation of cells that acquire mutations due to repetitive exposure to UV radiation, exogenous mutagens, and reactive oxygen
1306 Melanocytic nevi
A
stop proliferation. Furthermore, some cell cycle inhibitors are also found to be expressed in varying degrees in melanoma, indicating that the cells may have become resistant against their effects.18,19 The CDKN2A gene, which encodes p16 and p14, appears to play a particularly important role in melanocytic neoplasia. Chromosome 9p21, where CDKN2A resides, is the most frequent area of chromosomal loss in melanoma, and a recent study has shown that homozygous loss of CDKN2A occurs frequently during the transition from nevus to invasive melanoma.20
According to this concept of ‘oncogene-induced senescence’, the growth arrest of a cell that has acquired an oncogenic mutation presents itself as a relatively acute phenomenon. However, the cell cycle arrest found in nevi appears to occur with considerable latency. In order to be clinically detectable, nevi must consist of thousands of cells produced from a considerable number of cell divisions from a progenitor cell. Oncogenic mutations in genes such as BRAF are found in the majority of nevus cells, indicating that these cells must have proliferated in the presence of the mutation and propagated it through the population.21 The mechanism of oncogene-induced senescence outlined above, which relies on detecting oncogene activation by immediately triggering growth arrest in the form of a tripped fuse, does not explain this latency. It is possible that this immediate-type senescence exists in vivo, but as cells with oncogenic mutations would be arrested either immediately or after just a few rounds of divisions, the ensuing lesions would be clinically undetectable. This raises the intriguing possibility that in patients with a propensity to develop many nevi, oncogene-induced senescence may not be fully functional. Additional mechanisms of senescence appear to come into play if one of the barriers fails. For example, patients who are homozygous for loss-of-function mutations in the CDKN2A gene still develop nevi, although these nevi are typically large in size.22 Different mechanisms outside the p16/p14 pathways are responsible for growth arrest in such nevi.
B
species. For instance, constitutive activation of signaling pathways such as the MAP-kinase pathway by mutations in signaling components results in abnormally high constitutive signaling flux that differs from the signaling pattern induced by exposure to a growth factor.14 Under physiological conditions, growth signals attenuate rapidly through complex feedback mechanisms that dampen the input signal by, for example, receptor internalization and upregulation of inhibitory factors of critical signaling components (Fig. 25.260A). By contrast, excessive pathway activation by oncogenic mutations is thought to trigger similar and perhaps additional feedback mechanisms that counteract the growth-promoting effects (Fig. 25.260B). This involves the induction of cell cycle inhibitors such as p16 and p21 as well as other sensors of ‘oncogenic stress’ such as p14/ARF.15 Whereas p16 and p21 are inhibitors of cyclin-dependent kinases which inactivate the RB tumor suppressor, p14/ARF acts upstream of p53 and blocks its degradation.16 Oncogene activation engages the RB and p53 pathways which restrains proliferation. Expression of the above cell cycle inhibitory proteins can be detected in most nevi. Remarkably, however, the pattern of, for example, p16 expression within a nevus can be patchy, with some neoplastic melanocytes expressing robust levels, whereas others do not.17 This could indicate that not all cells within a lesion rely on the same factor or mechanism to
An alternative mechanism in the growth arrest of nevi is replicative or telomere-induced senescence (Fig. 25.260C). Telomeres are complex structures at chromosome ends that sequester the open ends of the DNA by forming a lariat structure with tandem DNA repeats protected by a matrix of nucleoproteins.23 In most somatic cells, telomeres shorten with each cell division and after 60–70 divisions reach a critical length that exposes the open end of the DNA and triggers a DNA damage signal that permanently arrests the cell.24 If this critical signal cascade is impaired, cells can continue to replicate and form end-to-end fusions of open telomeres between sister chromatids resulting in bicentric chromosomes. This leads to chromosome
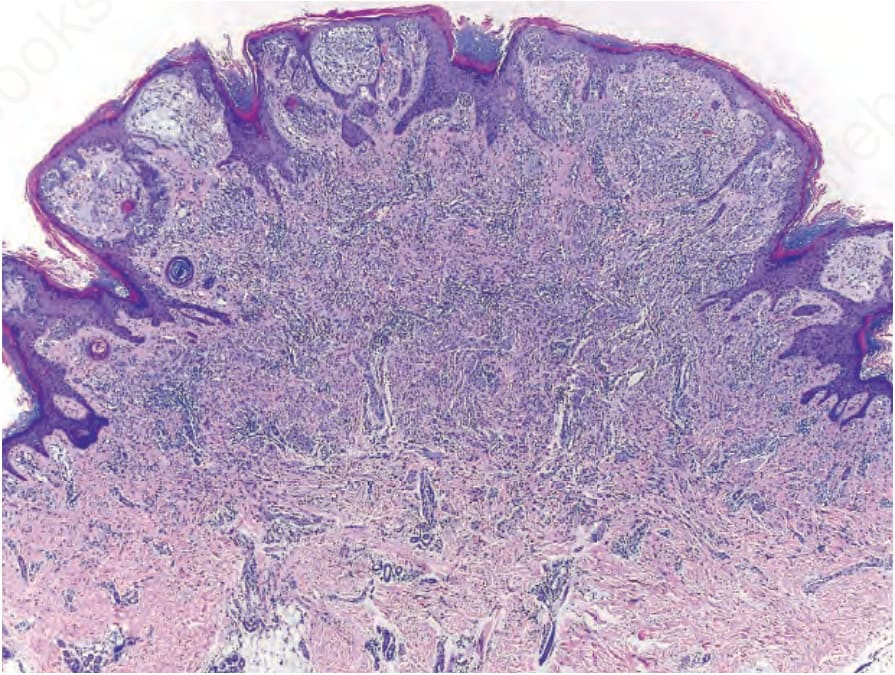
Fig. 25.255 Compound Spitz nevus with an ALK fusion: nests of melanocytes with clefts are present below an irregularly hyperplastic epidermis. Nests and fascicles of melanocytes stream from the junction into the reticular dermis with diminution of nest size in the deep portion.
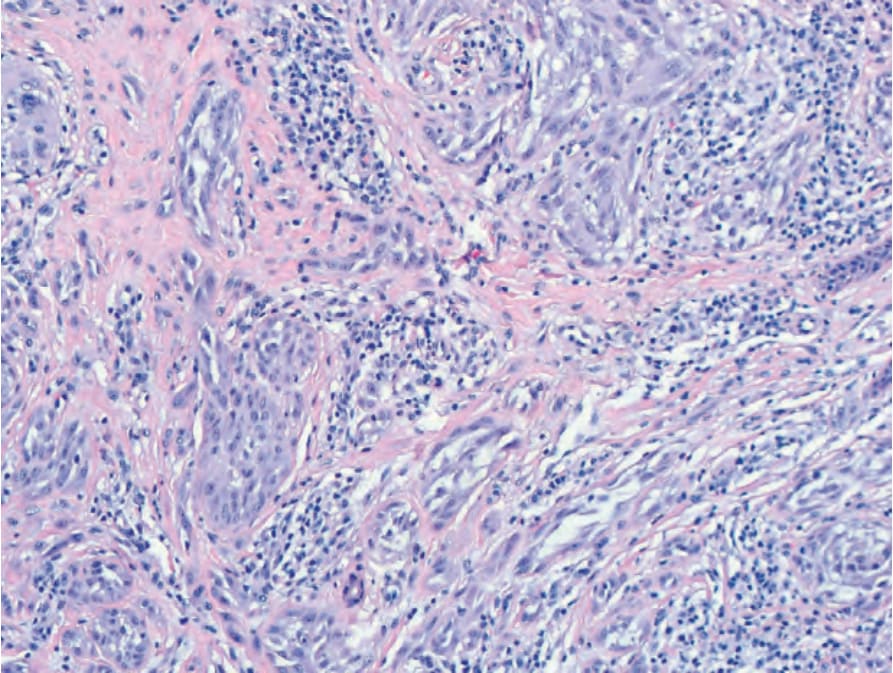
Fig. 25.256 Spitz nevus with an ALK fusion: fascicles of epithelioid and spindled melanocytes with prominent clefting in vertical orientation in the reticular dermis.
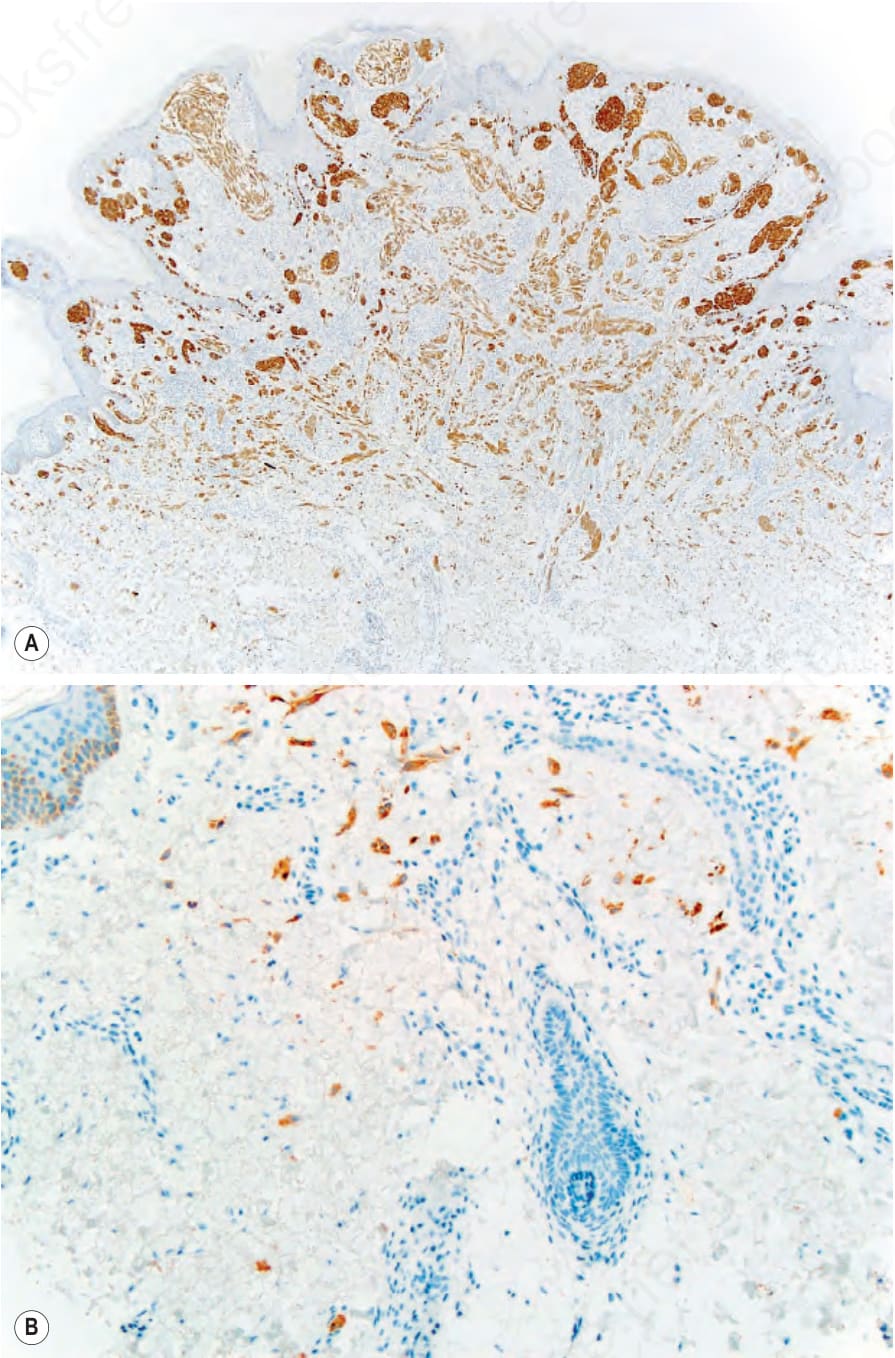
Fig. 25.257 (A) Spitz nevus with an ALK fusion: ALK immunostaining highlights the entire neoplasm, including an infiltrative pattern at the periphery, with single positive melanocytes extending far from the main portion of the neoplasm (B or inset).
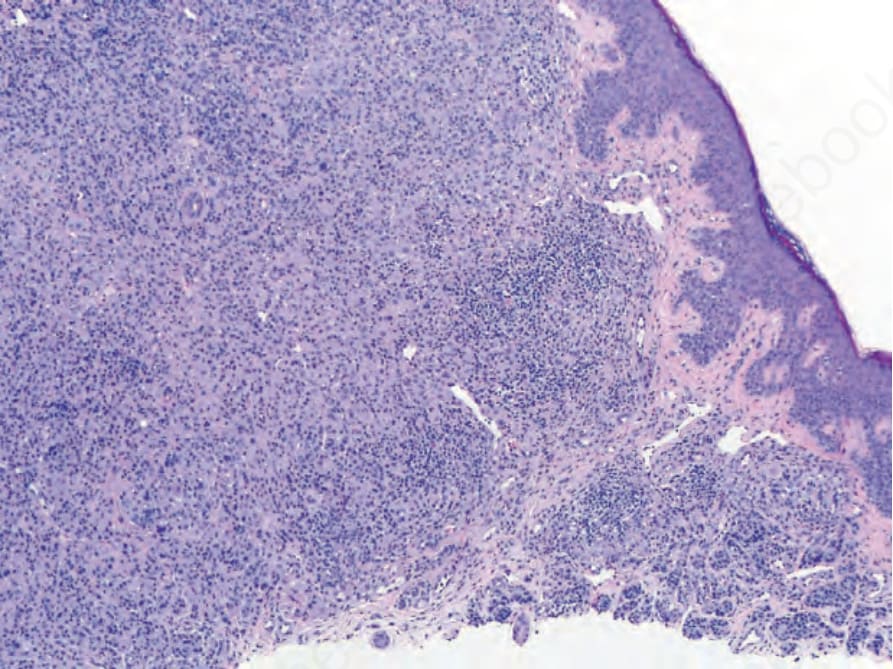
Fig. 25.258 BAP1-inactivated spitzoid tumor: adjacent to the large nodular collection of spitzoid cells, there are nests of small, round melanocytes representing a remnant population of the precursor nevus that retains a functional BAP1 protein (positive nuclear BAP1 staining).
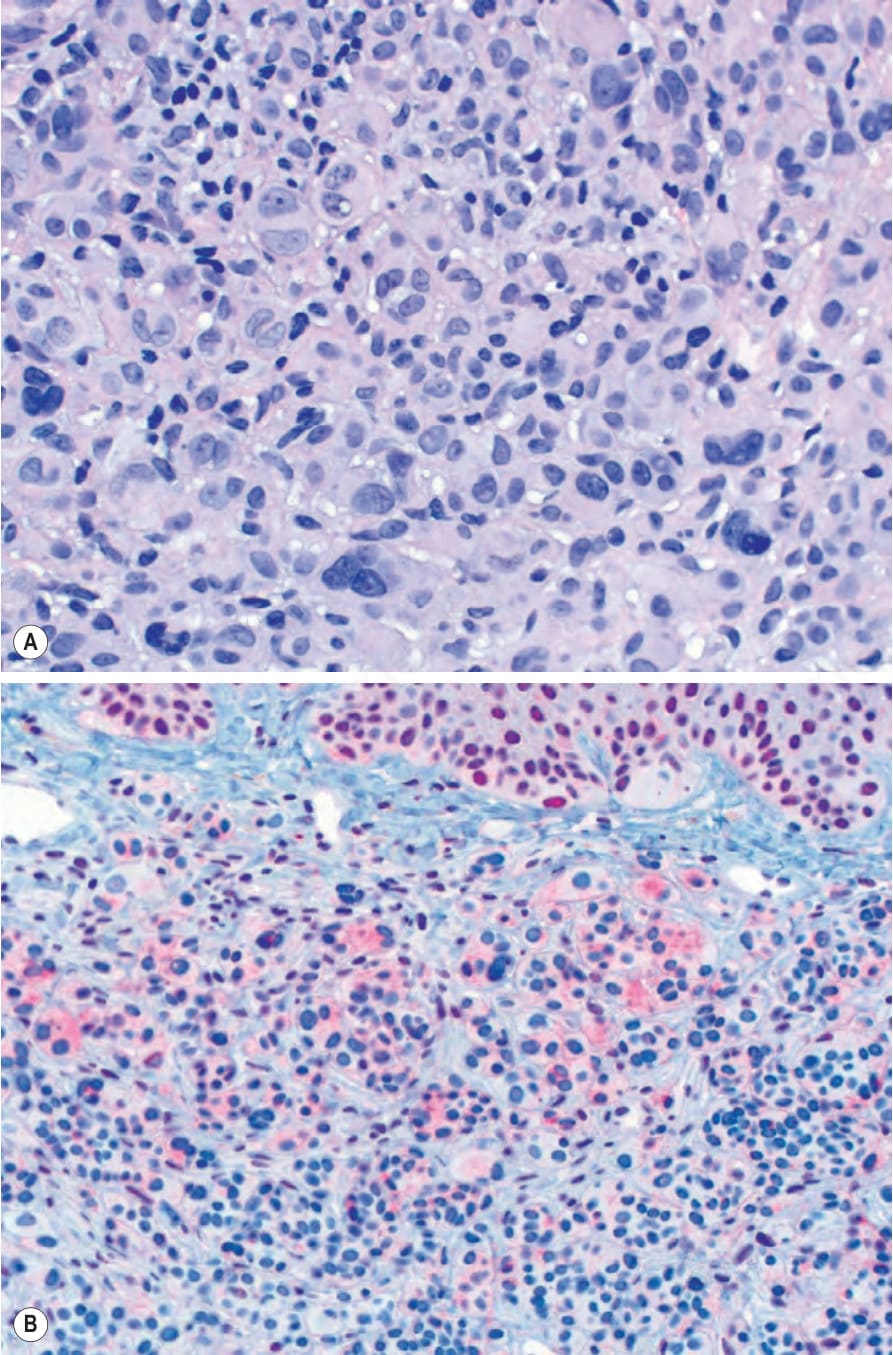
Fig. 25.259 (A) BAP1-inactivated spitzoid tumor: large epithelioid melanocytes with abundant pale cytoplasm form nodules in the dermis. Numerous multinucleated melanocytes are present. (B, inset) BAP1 stain showing intact nuclear staining of overlying epidermal keratinocytes and loss of nuclear staining in epithelioid melanocytes.
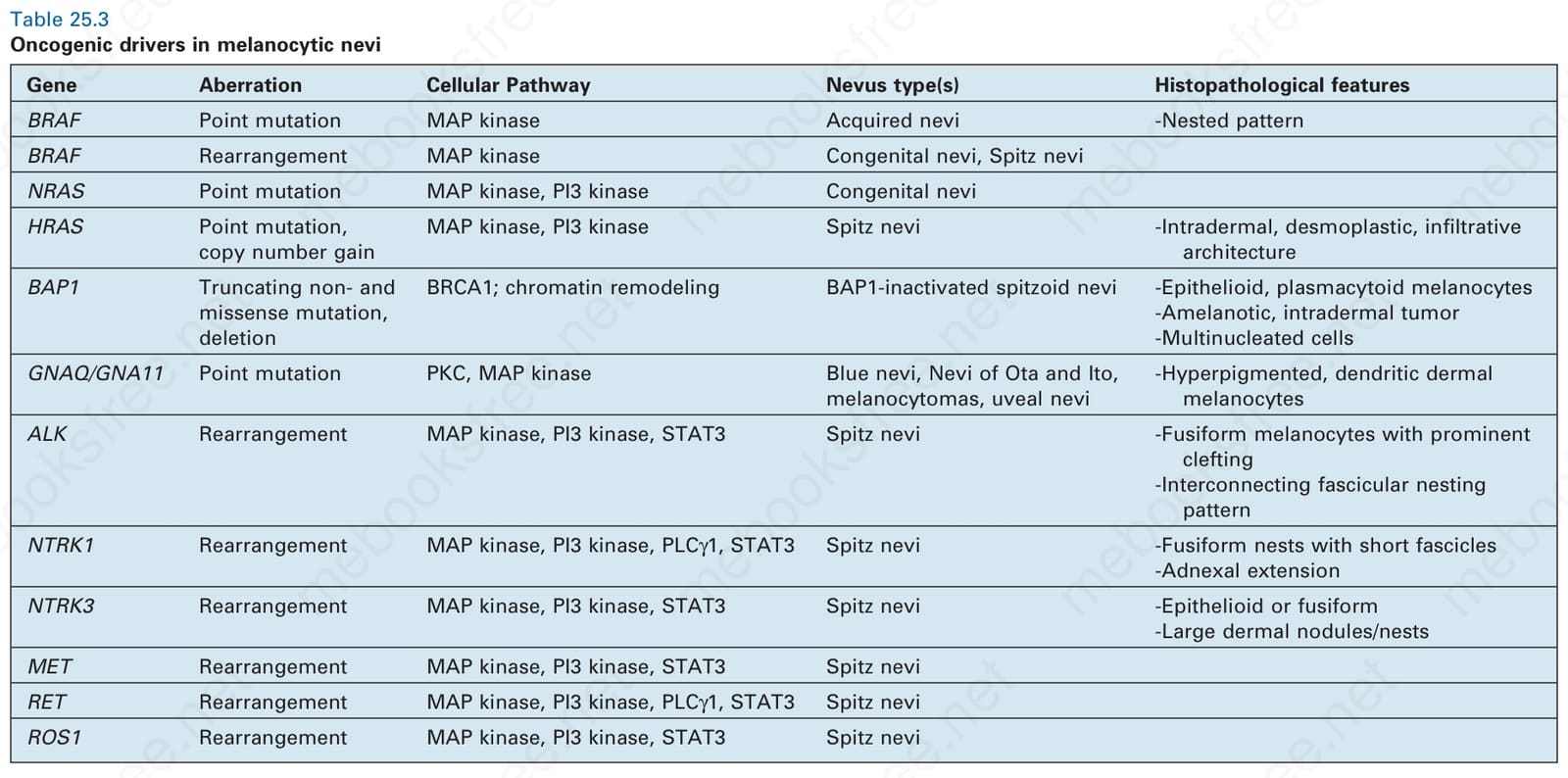
Table 25.3 Oncogenic drivers in melanocytic nevi
1307 The molecular pathology of melanocytic nevi
Growth signal
Growth signal
Receptor
Receptor
RAS
RAS
RAF
RAF
MEK
MEK
A
ERK
ERK
ETS
ETS
S-phase p16 A B
S-phase
60 divisions
C D
B
breakage/fusion/bridge cycles causing chromosomal gains and deletions. The ensuing genomic chaos results in a high frequency of cell death and is termed crisis.25 Crisis, on one hand, acts as a tumor preventative mechanism for cells that have become genetically unfit because they have suffered too many genetic alterations to be compatible with viability.24 On the other hand, it produces new genetic variants, some of which may have acquired increased proliferative capacity. The emergence of transformed clones occurs from rare cells that succeed in escaping crisis by acquiring the ability of restabilizing their telomeres, e.g., by activation of telomerase, an enzyme which extends telomeres using its own RNA template.26 Mutations in the TERT promoter can facilitate production of telomerase to overcome telomere-induced senescence and are found in numerous cancers, including melanoma.27 Recent study has shown that TERT promoter mutations are detectable already in intermediate-stage melanocytic neoplasms including dysplastic nevi, before full malignant transformation into melanoma has occurred.20 The presence of TERT activation this early in the evolution of melanocytic neoplasia indicates that the cells of nevi may continue to divide to the point which their telomeres are exhausted. Most nevi are
composed of comparatively small numbers of cells, estimated to be between 105 and 106 cells, much lower than the number expected at a replicative limit of 60 cell divisions. This marked discrepancy between the expected and observed number indicates very significant attritional factors that must eliminate many of the cells in the nevus. Elimination of cells by the immune system and/or cell-autonomous mechanisms such as apoptosis are likely candidates.
Such a dynamic scenario of nevus formation and maintenance, as opposed to that of a stable population of senescent cells, would explain the changes in nevus appearance in longitudinal studies of nevus appearance.28 It would also explain why nevi tend to fade after the age of 30. Once cells have reached their replicative life span, the balance between proliferation and attrition would tip in favor of attrition. Only lesions that have overcome this barrier by mutating the TERT promoter would be able to continue to proliferate (and acquire additional mutations to evolve towards melanoma).
Replicative life span is linked to nevus phenotype. Nevi acquired after birth typically do not exceed a size of 2 cm, whereas nevi that develop in utero when telomeres are longer can reach significantly larger sizes. This suggests that telomere size in the initiating melanocyte impacts the maximum size of the ensuing nevus,29 and studies on nevus size and density have found an association with telomere length.30 Additional mechanisms of senescence have been shown in nevi and colon adenomas that rely on an oncogene-induced inflammatory response involving interleukins 6 and 8.31
1308 Melanocytic nevi
The link between senescence and inflammation is interesting, because many nevi, in particular dysplastic nevi, show a chronic inflammatory infiltrate.
DNA damage can also trigger growth arrest. Mutations in oncogenes such as RAS family members lead to increased DNA replication errors resulting in DNA damage.32 Misfiring of replication origins results in abnormal DNA structures that trigger critical checkpoints, typically in a p53-dependent manner, which arrest the cells and permanently stop proliferation if the issue cannot be resolved by DNA repair mechanisms (see Fig. 25.260). If the problem cannot be repaired, the cells undergo a permanent growth arrest. This type of senescence has been termed DNA damaged-induced senescence.33,34 The mechanism has been demonstrated in several types of precancerous lesions, including melanocytic nevi.33 The scenario of DNA damage-induced senescence implies that a nevus can harbor cells with genetic alterations beyond simple point mutations, which arose from replication errors or missegregation of chromosomes. However, the induction of senescence in these cells restricts them from clonal expansion (Fig. 25.261A). By contrast, in melanoma these checkpoints are lost, allowing the proliferation of melanocytes with an abnormal genome from which clones with a favorable genomic constellation become selected
(Fig. 25.261B). This scenario would explain why clonal chromosomal aberrations are mostly absent in nevi but are very common in melanoma (Fig. 25.262).
In summary, current evidence strongly suggests that several independent mechanisms restrain the proliferation of melanocytes that have been initiated by activating mutations in potent oncogenes and, in concert, provide a robust barrier to cancer formation. Even if one mechanism fails, backup mechanisms can prevent malignant transformation. This concept would explain why individuals with loss-of-function alleles of CDKN2A can still develop nevi, albeit of larger sizes. However, if one of the senescence mechanisms is impaired, an increased risk for melanoma could ensue, explaining why inherited mutations in CDKN2A are a strong risk factor for melanoma.

Fig. 25.260 Examples of mechanisms of senescence. (A) Under physiological circumstances, a growth signal is relayed down the MAP-kinase pathway and in the absence of inhibitory signals results in cell division. (B) If the pathway is hyperactivated, for example, by a mutation at the level of RAS, mediators of senescence such as p16 become induced and inhibit cell cycle entry. (C) Replicative senescence ensues when a population of cells continues to divide and exhausts its replicative potential due to telomere erosion. (D) DNA damaged-induced senescence is caused by random genetic alterations during the cell cycle with subsequent permanent growth arrest or apoptosis.
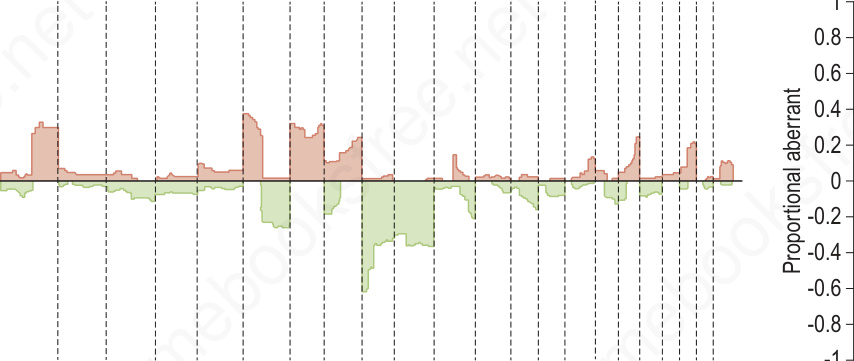
Fig. 25.262 Comparative genomic hybridization of melanoma (malignant) and nevi (benign): on the left, melanomas (n = 133) are associated with multiple copy number gains and losses that cluster. In contrast, the nevi (n = 54) on the right show minimal changes with the exception of the 11p copy number increase or amplification in Spitz nevi which includes the HRAS gene. These differences can be exploited using multiplexed FISH assays to support the diagnosis of melanoma or nevus in challenging cases.