McCune-Albright 症候群 (McCune-Albright syndrome)
臨床特徵 (Clinical Features)
- McCune-Albright 症候群 (McCune-Albright syndrome)(亦稱為 Albright disease 或 polyostotic fibrous dysplasia)是一種罕見疾病,最初以咖啡牛奶斑 (café-au-lait macules)、多骨性纖維異常增生 (polyostotic fibrous dysplasia) 與性早熟 (sexual precocity)(主要發生於女性)為特徵。
- 在具備三項主要特徵中的兩項時,即可診斷本病。其他罕見表現包括甲狀腺機能亢進 (hyperthyroidism)、肝臟疾病、生長激素分泌過多 (growth hormone hypersecretion)、貧血 (anemia)、高泌乳素血症 (hyperprolactinemia)、Cushing disease、伴隨或不伴隨佝僂病/骨軟化症 (rickets/osteomalacia) 的腎臟磷酸鹽流失 (renal phosphate wasting),以及肝臟與心臟侵犯。
- 本病主要影響女性。皮膚與軟組織內可見骨瘤 (osteomas) 與鈣化。
- 皮膚的咖啡牛奶斑 (café-au-lait spots) 為不對稱分布,於出生後最初 2 年內出現,較少見的情況是出生時即已存在。其主要侵犯軀幹與下肢近端,較少見於臉部與頸部(Fig. 20.26)。
- 有人提出,McCune-Albright syndrome 中的色素斑可藉由咖啡牛奶斑邊緣較為平滑而與神經纖維瘤病 (neurofibromatosis) 所見的病灶區分(後者邊緣較平滑)。然而並非總是如此,鑑別診斷可能極為困難。
致病機轉與組織學特徵 (Pathogenesis and Histologic Features)
- 本症候群中已描述一種非遺傳性、合子後 (postzygotic) 的活化性突變,發生於由 GNAS 基因所編碼的 Gs alpha 蛋白。
- Gs alpha 蛋白是 G 蛋白複合體 (G-protein complex) 的組成成分,該複合體將荷爾蒙受體連接至腺苷酸環化酶 (adenylate cyclase)。此蛋白的活化會導致多種不同蛋白的過度產生。
- 來自色素性斑狀病灶的皮膚切片顯示基底細胞 (basal cells) 過度色素沉著。有時可見巨大黑色素小體 (giant melanosomes),但其出現頻率不如第一型神經纖維瘤病 (neurofibromatosis type I) 的咖啡牛奶斑中那麼常見。
- 在真皮與皮下組織中,可見斑塊狀骨瘤 (plaquelike osteomas)(Fig. 20.27)。罕見情況下,皮膚與軟組織中亦曾描述其他病灶,包括含骨樣組織的痣 (nevi with osteoid)、類鈣化性腱膜纖維瘤 (calcifying aponeurotic fibroma-like lesions) 病灶,以及類局限性鈣質沉著 (calcinosis circumscripta-like lesions) 病灶。
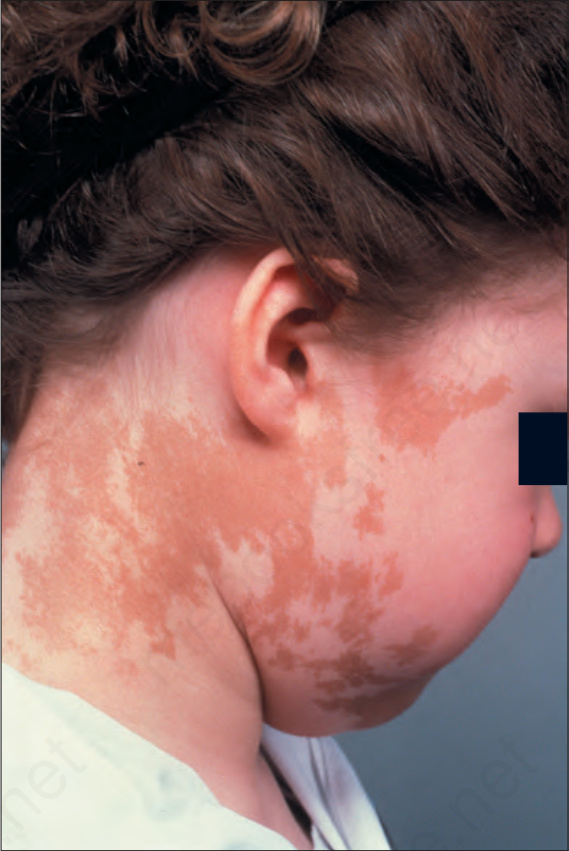
圖 20-26:McCune-Albright 症候群:邊緣不規則的淡棕色色素性斑狀病灶。By courtesy of the Institute of Dermatology, London, UK. (McCune-Albright syndrome)
Fig. 20.26 McCune-Albright syndrome: light-brown pigmented macular lesions with an irregular margin. By courtesy of the Institute of Dermatology, London, UK.
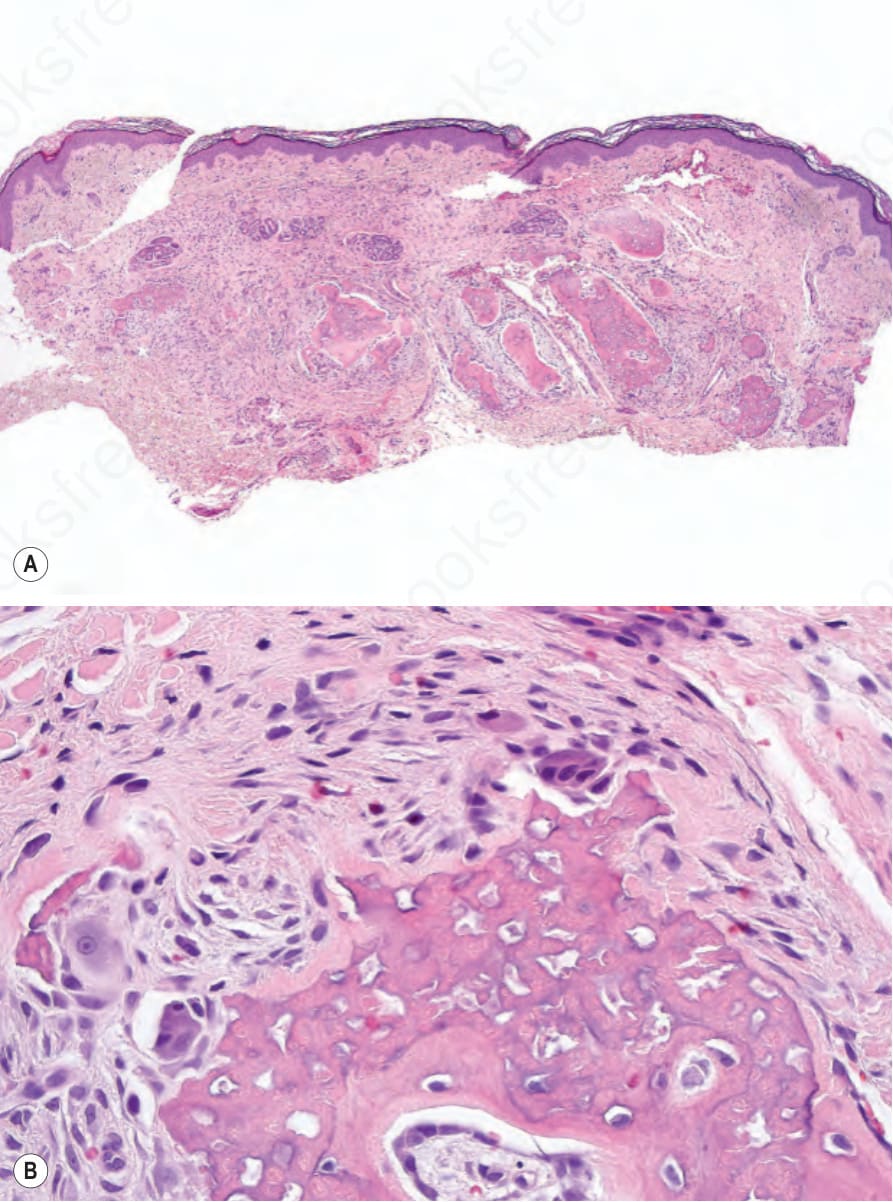
圖 20-27:McCune-Albright 症候群:真皮內可見骨樣組織 (A);高倍視野 (B)。 (McCune-Albright syndrome; osteoid)
Fig. 20.27 McCune-Albright syndrome: osteoid is present in the dermis (A); high-power view (B).
屈側網狀色素異常 (Dowling-Degos disease)
臨床特徵 (Clinical Features)
- 此罕見疾病(亦稱為 Dowling-Degos disease,DD disease)具有體染色體顯性 (autosomal dominant) 遺傳模式。
- 其表現為進行性演變的小型(1–2 mm)過度色素沉著斑點,呈網狀 (reticulate) 或融合性 (confluent) 分布,好發於屈側區域,包括腋窩 (axillae)、肘前窩 (antecubital fossae)、乳房下區 (inframammary regions)、頸部與腹股溝 (groins)(Fig. 20.28)。亦可見外陰部 (vulva) 受侵(有時為唯一表現)以及(更罕見地)頭皮受侵。
- 其他特徵包括小型、色素性的類粉刺病灶(暗點毛囊,dark dot follicles)、色素性毛囊性過度角化丘疹 (pigmented, follicular hyperkeratotic papules)(見 Fig. 20.28A),以及口周凹陷性痤瘡樣疤痕 (perioral pitted acneiform scars)。
- 有時可見色素減退或無色素 (hypopigmented or achromic) 斑。曾有一例報告呈現廣泛全身性過度色素沉著合併色素減退斑。
- 通常於青春期或成年早期表現。女性較常受影響。本病偶爾會因受熱而惡化。
- 罕見病患曾合併智能障礙 (mental retardation)。本病已描述的相關疾病包括化膿性汗腺炎 (hidradenitis suppurativa)、多發性表皮樣與毛根鞘囊腫 (multiple epidermoid and trichilemmal cysts)、關節炎 (arthritis)、多發性角化棘皮瘤 (multiple keratoacanthomas)、多發性脂漏性角化症 (multiple seborrheic keratoses),以及鱗狀細胞癌 (squamous cell carcinoma)。曾有一例記錄鱗狀細胞癌發生於色素沉著區域。亦曾描述一名病患同時呈現 DD disease 與 Darier disease,此可能屬巧合性表現。
- 多年來逐漸增加的證據顯示 DD disease 具有一系列不同的表型表現,包括 Galli-Galli disease(見下文)。在 Galli-Galli disease 中,其表現與典型 DD disease 完全相同,但組織學上可見基底上層非角化不良性棘層鬆解 (suprabasal nondyskeratotic acantholysis)。曾描述一種 Galli-Galli disease 的變異型,其缺乏網狀過度色素沉著,而以軀幹及下肢的紅斑性鱗屑斑塊與類雀斑樣斑 (lentigo-like macules) 表現。
致病機轉與組織學特徵 (Pathogenesis and Histologic Features)
- DD disease 的表型譜系可歸因於三個對黑色素運輸 (melanin transport) 至為關鍵之基因的功能喪失型突變 (loss-of-function mutations)。這三個基因為 KRT5(編碼 keratin 5)、POFUT1(編碼 protein O-fucosyltransferase)與 POGLUT1(編碼 protein O-glucosyltransferase)。這些突變可能導致角質形成細胞 (keratinocytes) 內黑色素小體 (melanosomes) 分布的改變。
- KRT5 突變主要導致出現於年輕成人的屈側型疾病。POFUT1 突變導致類似但更廣泛的疾病,不僅侵犯屈側,亦侵犯軀幹、腹部、頸部與肢端部位;亦可見掌部凹陷 (palmar pits) 與中斷的指紋皮紋 (interrupted dermatoglyphs)。POGLUT1 突變所導致的疾病可能在生命早期或較晚期表現,主要與非屈側區域(包括軀幹與四肢)的侵犯相關,皮膚切片可能呈現局灶性棘層鬆解 (focal acantholysis)。
- 一種以化膿性汗腺炎 (hidradenitis suppurativa,acne inversa) 表現的 DD disease 變異型,與編碼 γ-secretase 次單元的 PSENEN(編碼 presenilin enhancer protein 2)基因突變相關。
- 其特徵令人聯想到腺樣型脂漏性角化症 (adenoid seborrheic keratosis)。在厚度正常或減少的表皮上方可見過度角化 (hyperkeratosis)。細長、絲狀、色素性、相互連接的上皮索 (epithelial strands) 自表皮與毛囊壁向下生長進入淺層真皮(Fig. 20.29)。毛囊的漏斗部 (infundibular portion) 呈現擴張。偶可見小型角質囊腫 (horn cysts)。發炎通常不是特徵,但可能出現苔癬樣組織反應 (lichenoid tissue reaction)。在 Galli-Galli disease 中,可見局灶性棘層鬆解 (focal acantholysis)(Fig. 20.30)。
- 超微結構研究顯示黑色素細胞 (melanocytes) 數目正常,但部分與完全黑色素化的黑色素小體 (melanosomes) 數目增加。角質形成細胞 (keratinocytes) 含有分散的單一黑色素小體,這與白人皮膚中通常所見的複合聚集體 (compound aggregates) 形成對比。
鑑別診斷 (Differential Diagnosis)
- 與腺樣型脂漏性角化症 (adenoid seborrheic keratosis) 的區分可能有困難,但 DD disease 中所見的絲狀上皮索 (filiform epithelial strands) 與毛囊的漏斗部 (infundibular portion) 關係非常密切。臨床背景對於達成診斷非常有幫助。
- 上述組織學變化頗具特色,但對本病並非具特異性。Haber syndrome 以類酒糟 (rosacea-like) 的顏面疹、凹陷性疤痕 (pitted scars),以及軀幹與屈側的類脂漏性角化症 (seborrheic keratoses-like) 病灶為特徵。其他已報告的特徵包括掌蹠角化症 (palmoplantar keratoderma) 與明顯的指甲表皮 (prominent nail cuticles)。雖然有人提出 Haber syndrome 是 DD disease 的一種變異型,但其臨床表現不同,且迄今尚未發現 keratin 5 的突變。
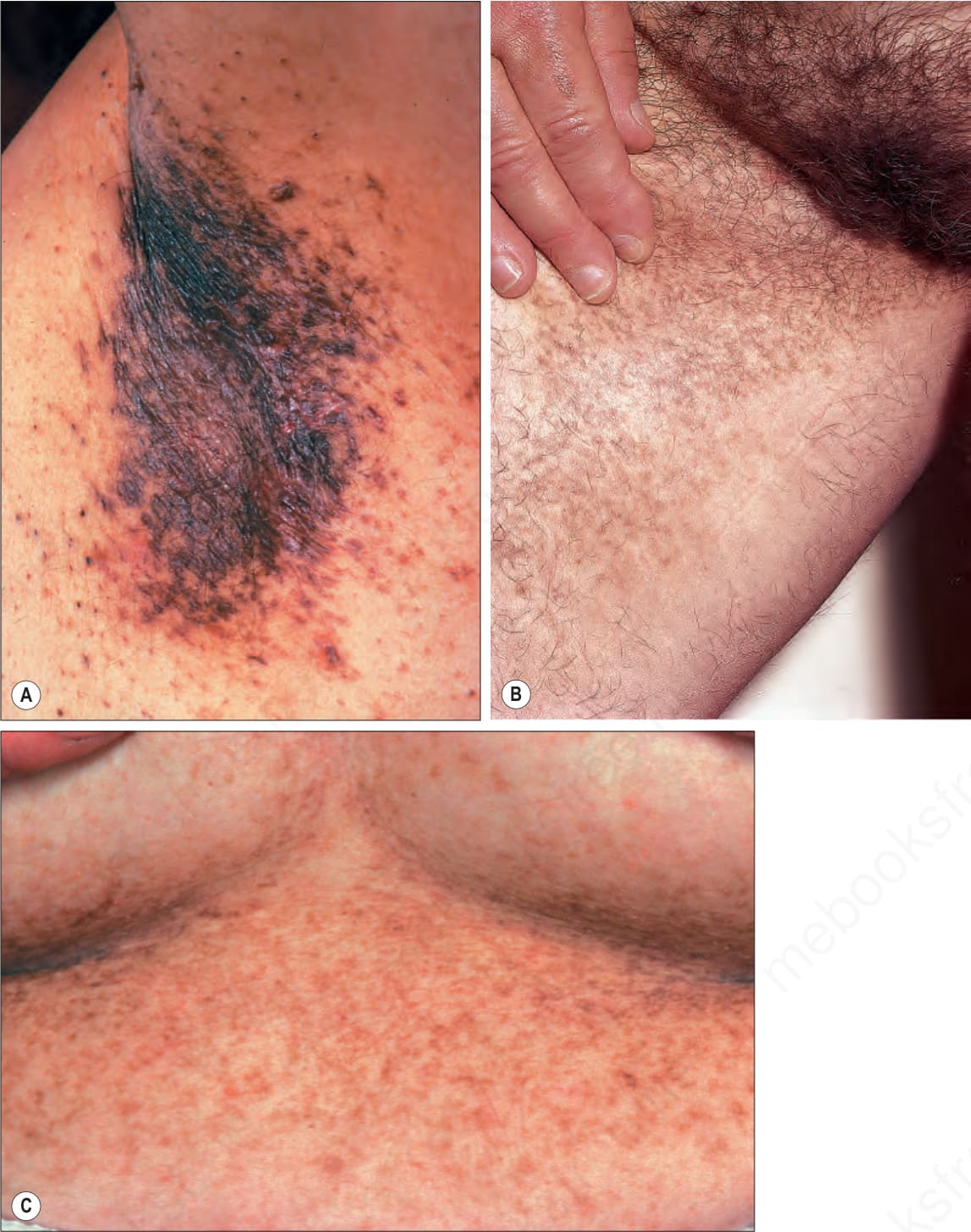
圖 20-28:Dowling-Degos disease:色素性網狀斑出現於 (A) 腋窩、(B) 腹股溝與 (C) 乳房下區。亦請注意腋窩上的毛囊性色素性過度角化丘疹。(A, C) By courtesy of the Institute of Dermatology, London, UK;(B) by courtesy of E. Wilson Jones, MD, Institute of Dermatology, London, UK. (Dowling-Degos disease)
Fig. 20.28 Dowling-Degos disease: pigmented reticulate macules on (A) the axilla, (B) the groin, and (C) the inframammary area. Note also follicular pigmented hyperkeratotic papules on the axilla. (A, C) By courtesy of the Institute of Dermatology, London, UK; (B) by courtesy of E. Wilson Jones, MD, Institute of Dermatology, London, UK.
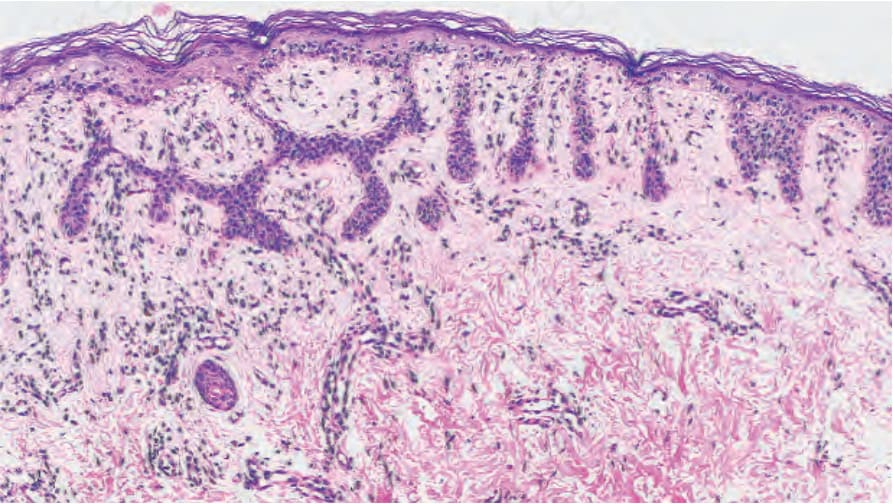
圖 20-29:Dowling-Degos disease:請注意過度色素沉著的細薄表皮索。By courtesy of E. Wilson Jones, MD, Institute of Dermatology, London, UK. (Dowling-Degos disease)
Fig. 20.29 Dowling-Degos disease: note the hyperpigmented thin epidermal strands. By courtesy of E. Wilson Jones, MD, Institute of Dermatology, London, UK.
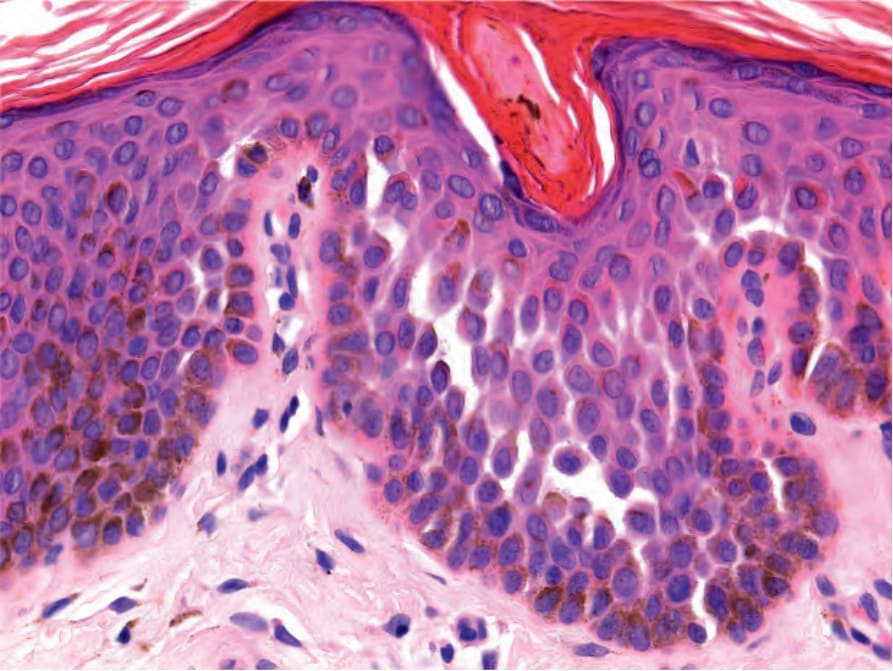
圖 20-30:Dowling-Degos disease 的 Galli-Galli 變異型:可見局灶性非角化不良性棘層鬆解。 (Galli-Galli variant of Dowling-Degos disease; nondyskeratotic acantholysis)
Fig. 20.30 Galli-Galli variant of Dowling-Degos disease: focal nondyskeratotic acantholysis is seen.
- 在這些病例中,可見蹠部角化症 (plantar keratoderma)(Figs 20.31 and 20.32A & B)。本病的進展於中年時停止。
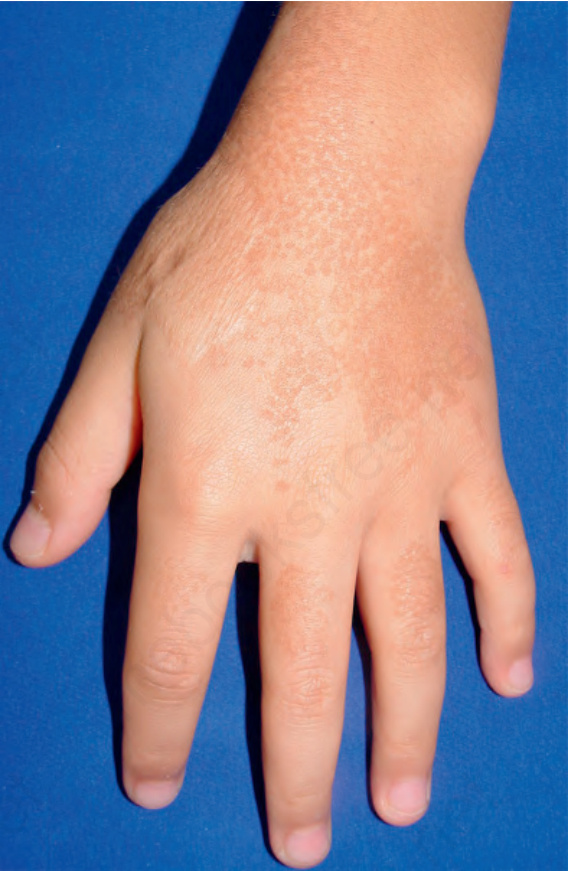
圖 20-31:Kitamura 網狀肢端色素沉著症 (macular reticulate pigmentation)。By courtesy of Dr. Chao Sheau-Chiou, Tainan, Taiwan. (Reticulate acropigmentation of Kitamura)
Fig. 20.31 Reticulate acropigmentation of Kitamura (macular reticulate pigmentation). By courtesy of Dr. Chao Sheau-Chiou, Tainan, Taiwan.