McCune-Albright syndrome
McCune-Albright syndrome
Clinical features McCune-Albright syndrome (also known as Albright disease or polyostotic fibrous dysplasia) is a rare disease initially characterized by café-au-lait macules, polyostotic fibrous dysplasia, and sexual precocity (mainly in females).1,2 The disease can be diagnosed in the presence of two of the three major features. Other rare manifestations include hyperthyroidism,
liver disease, growth hormone hypersecretion, anemia, hyperprolactinemia, Cushing disease, renal phosphate wasting with or without rickets/ osteomalacia, and hepatic and cardiac involvement.3,4 It mainly affects females. Osteomas and calcifications in the skin and soft tissues may be seen.5,6 Cutaneous café-au-lait spots are asymmetrical and develop during the first 2 years of life or more infrequently are present at birth.7 They mainly involve the trunk and proximal lower limbs and less commonly the face and neck (Fig. 20.26). It has been proposed that the pigmented macules
1004 Disorders of pigmentation
evolution of small (1–2 mm) hyperpigmented macules in a reticulate or confluent distribution, showing a predilection for the flexural regions including axillae, antecubital fossae, inframammary regions, neck, and groins (Fig. 20.28).1–8 Involvement of the vulva (sometimes as an exclusive manifestation) and (more rarely) the scalp may also be seen.9–11 Small, pigmented comedone-like lesions (dark dot follicles), pigmented, follicular hyperkeratotic papules (see Fig. 20.28A), and perioral pitted acneiform scars are also features.3
Hypopigmented or achromic macules are sometimes seen.12,13 A case with extensive generalized hyperpigmentation and hypopigmented macules has been reported.14 Presentation is usually in adolescence or early adulthood.15 Females are more frequently affected.4 The disease may occasionally be aggravated by heat.16 Rare patients have had associated mental retardation. Described associations of the disease include, hidradenitis suppurativa, multiple epidermoid and trichilemmal cysts, arthritis, multiple keratoacanthomas, multiple seborrheic keratoses, and squamous cell carcinoma.2,3,17–22 A case of squamous cell carcinoma arising in an area of pigmentation has been documented.23 A likely coincidental presentation of DD disease and Darier disease in the same patient has been described.24
A
Increasing evidence over the years suggests a range of different phenotypic presentations in DD disease including Galli-Galli disease (see below). In Galli-Galli disease, the presentation is identical to that of classic DD disease but histologically there is suprabasal nondyskeratotic acantholysis.25–28 A variant of Galli-Galli disease lacking reticulate hyperpigmentation and presenting with erythematous scaly plaques and lentigo-like macules of the trunk and lower limbs has been described.29
B
Pathogenesis and histologic features The phenotypic spectrum of DD disease can be attributed to loss-of-function mutations in three genes that are pivotal in melanin transport. These are KRT5 (encoding keratin 5), POFUT1 (encoding protein O-fucosyltransferase), and POGLUT1 (encoding protein O-glucosyltransferase).30–34 These mutations likely result in alteration in the distribution of melanosomes within keratinocytes. Mutations in KRT5 result mainly in flexural disease appearing in young adults.35 Mutations in POFUT1 result a similar but more generalized disease with involvement not only of the flexures, but also the trunk, abdomen, neck, and acral sites.35 Palmar pits and interrupted dermatoglyphs may also be seen. Mutations in POGLUT1 result in a disease that may be expressed early or later in life and is associated mainly with involvement of non-flexural areas, including trunk and extremities, and skin biopsies may display focal acantholysis.35,36 A variant of DD disease presenting with hidradenitis suppurativa (acne inversa) is associated with mutations in the gamma-secretase subunit-encoding PSENEN (encoding presenilin enhancer protein 2).37
in McCune-Albright syndrome may be distinguished from the lesions seen in neurofibromatosis by a smoother margin in the café-au-lait spots in the latter. However, this is not always the case and differential diagnosis may be very difficult.
Pathogenesis and histologic features A noninherited postzygotic activating mutation in the Gs alpha protein encoded by the GNAS gene has been described in the syndrome.4,8–10 The Gs alpha protein is a component of the G-protein complex that binds hormone receptors to adenylate cyclase. The activation of this protein results in overproduction of different proteins.
The features are reminiscent of an adenoid seborrheic keratosis. There is hyperkeratosis overlying an epidermis of normal or reduced thickness. Thin, filiform, pigmented, interconnecting epithelial strands grow down from the epidermis and walls of hair follicles into the superficial dermis (Fig. 20.29).15,38 The infundibular portion of the hair follicles appears dilated. Occasional small horn cysts may be present. Inflammation is not usually a feature but a lichenoid tissue reaction may be present. In Galli-Galli disease, focal acantholysis is seen (Fig. 20.30)
A skin biopsy from a pigmented macular lesion shows hyperpigmentation of basal cells. Giant melanosomes are sometimes seen but are not as frequent as in the café-au-lait spots of neurofibromatosis type I. In the dermis and subcutaneous tissue, plaquelike osteomas may be seen (Fig. 20.27). Rarely, other lesions have been described in skin and soft tissues including nevi with osteoid, calcifying aponeurotic fibroma-like lesions, and calcinosis circumscripta-like lesions.6
Reticulate pigmented anomaly of the flexures (Dowling-Degos disease)
Clinical features This rare condition (also known as Dowling-Degos disease, DD disease) has an autosomal dominant mode of inheritance. It presents with a progressive
Ultrastructural studies have shown a normal number of melanocytes with increased numbers of partially and fully melanized melanosomes.38,39 The keratinocytes contain dispersed single melanosomes in contrast to the compound aggregates usually seen in white skin.38,39
Differential diagnosis Distinction from an adenoid seborrheic keratosis may be difficult but the filiform epithelial strands seen in DD disease are very closely related to the infundibular portion of the hair follicles. The clinical background is very helpful in reaching a diagnosis.
The histologic changes are quite distinctive but not specific of the disease. Haber syndrome is characterized by a facial rosacea-like eruption, pitted scars, and seborrheic keratoses-like lesions on the trunk and flexures.40–44 Additional reported features include palmoplantar keratoderma and prominent nail cuticles.45 Although it has been suggested that Haber syndrome is
1005 Reticulate acropigmentation of Kitamura
A
B
C
a variant of DD disease, the clinical presentation is different and mutations in keratin 5 have not been found so far.46
cases, plantar keratoderma (Figs 20.31 and 20.32A & B).2,8,9 Progression of the disease stops in middle age.
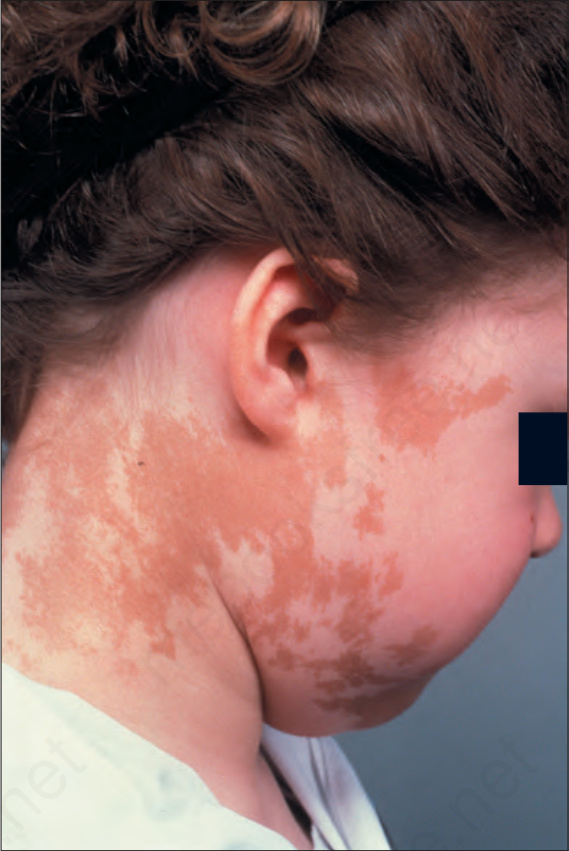
Fig. 20.26 McCune-Albright syndrome: light-brown pigmented macular lesions with an irregular margin. By courtesy of the Institute of Dermatology, London, UK.
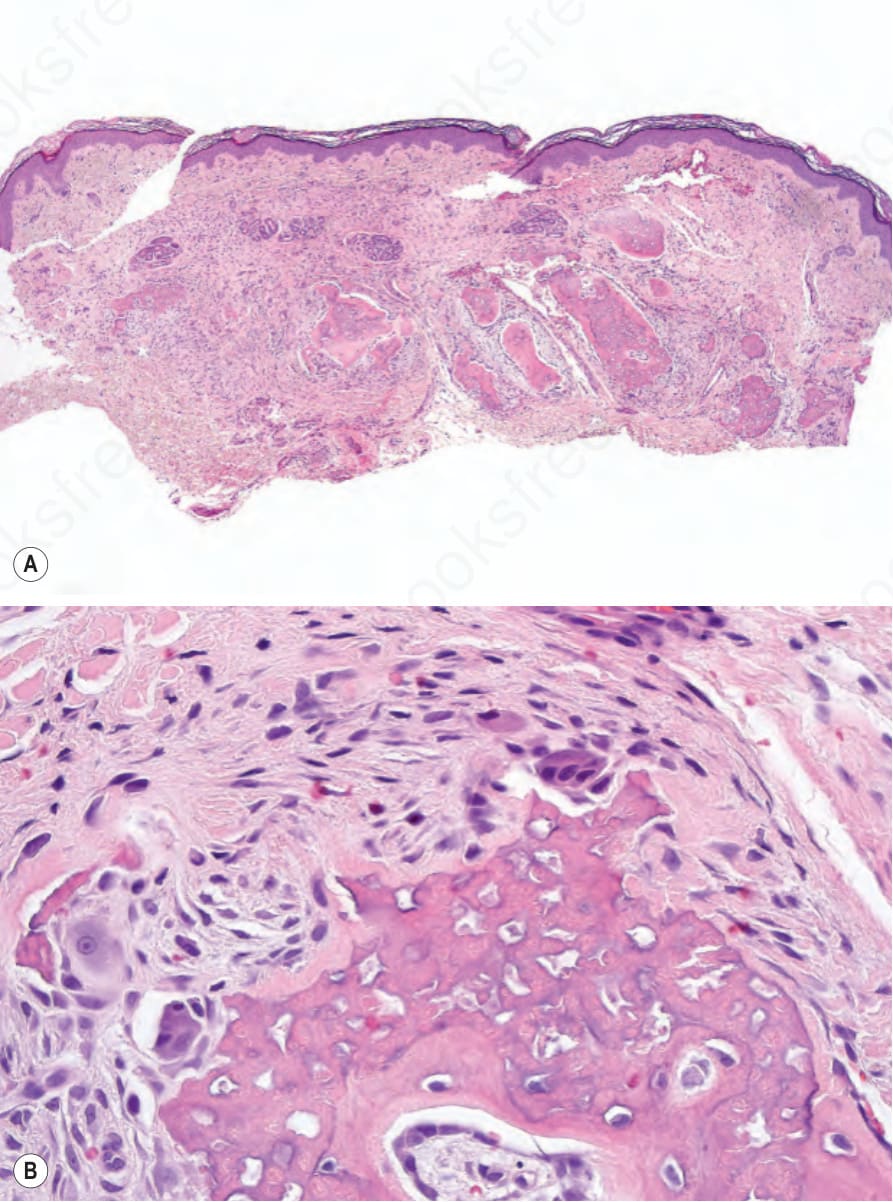
Fig. 20.27 McCune-Albright syndrome: osteoid is present in the dermis (A); high-power view (B).
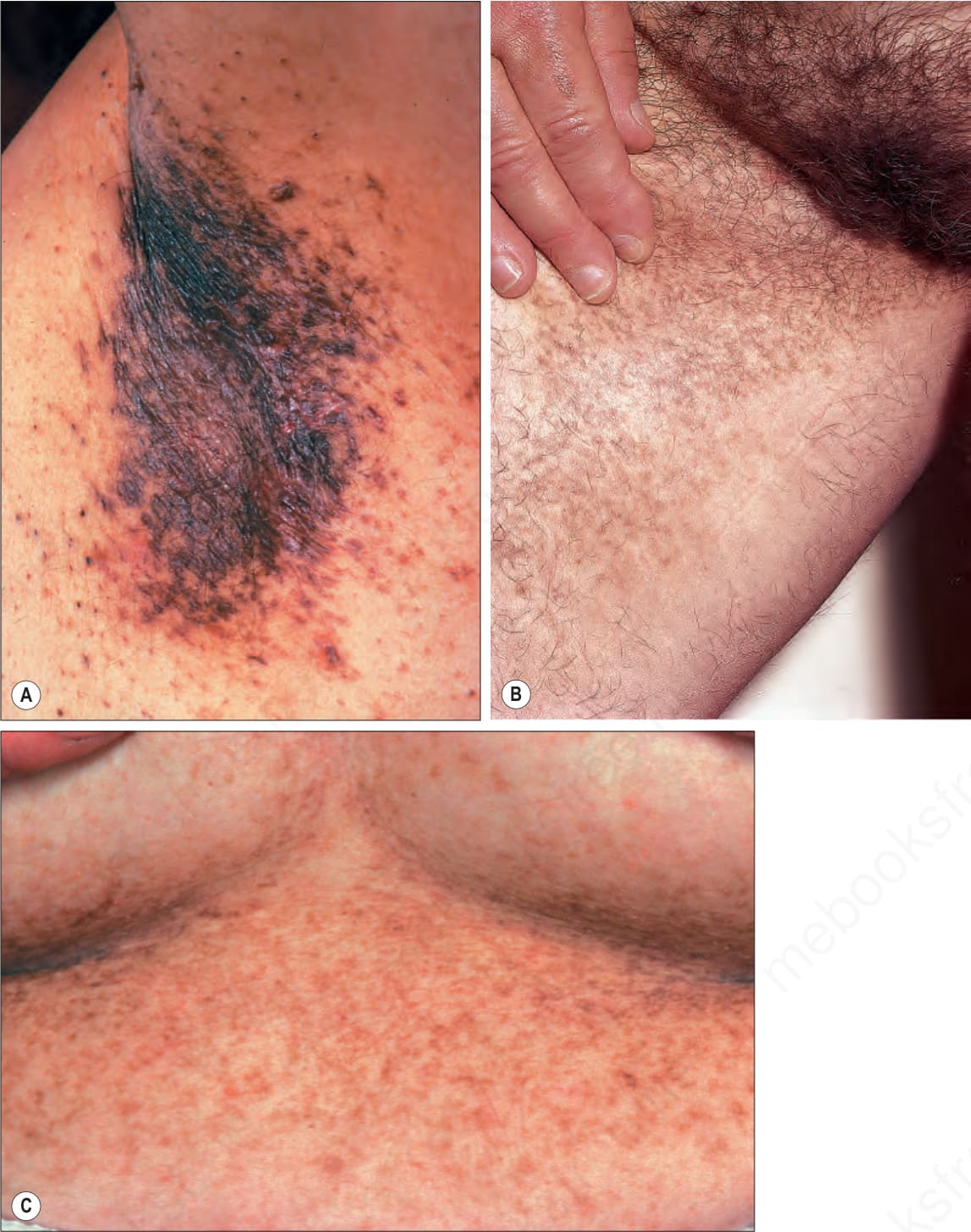
Fig. 20.28 Dowling-Degos disease: pigmented reticulate macules on (A) the axilla, (B) the groin, and (C) the inframammary area. Note also follicular pigmented hyperkeratotic papules on the axilla. (A, C) By courtesy of the Institute of Dermatology, London, UK; (B) by courtesy of E. Wilson Jones, MD, Institute of Dermatology, London, UK.
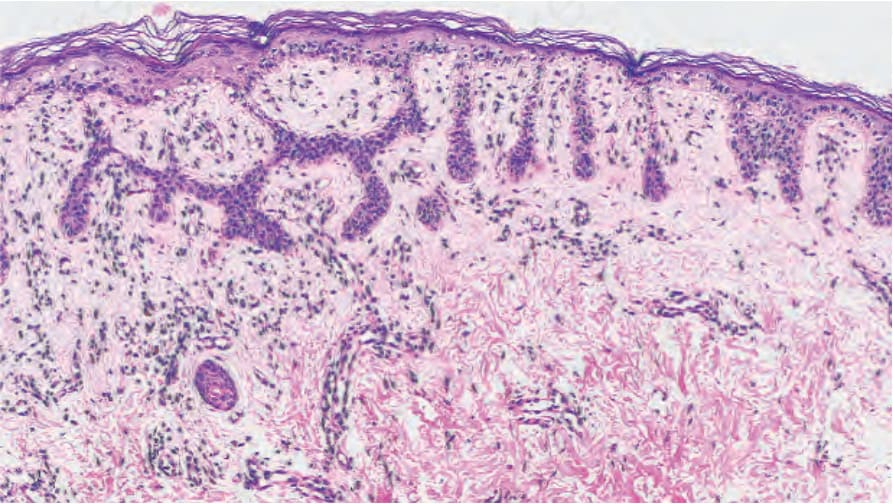
Fig. 20.29 Dowling-Degos disease: note the hyperpigmented thin epidermal strands. By courtesy of E. Wilson Jones, MD, Institute of Dermatology, London, UK.
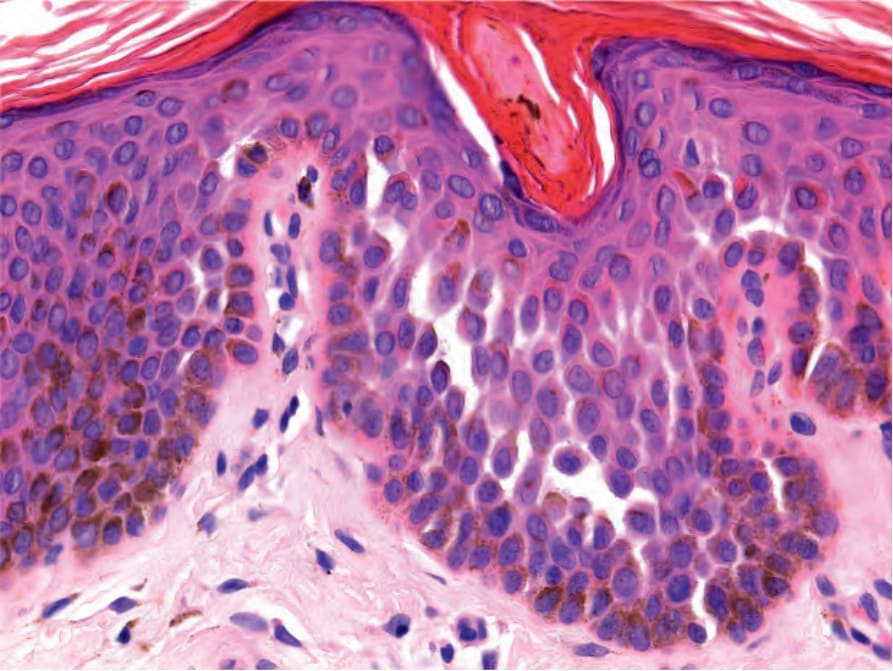
Fig. 20.30 Galli-Galli variant of Dowling-Degos disease: focal nondyskeratotic acantholysis is seen.
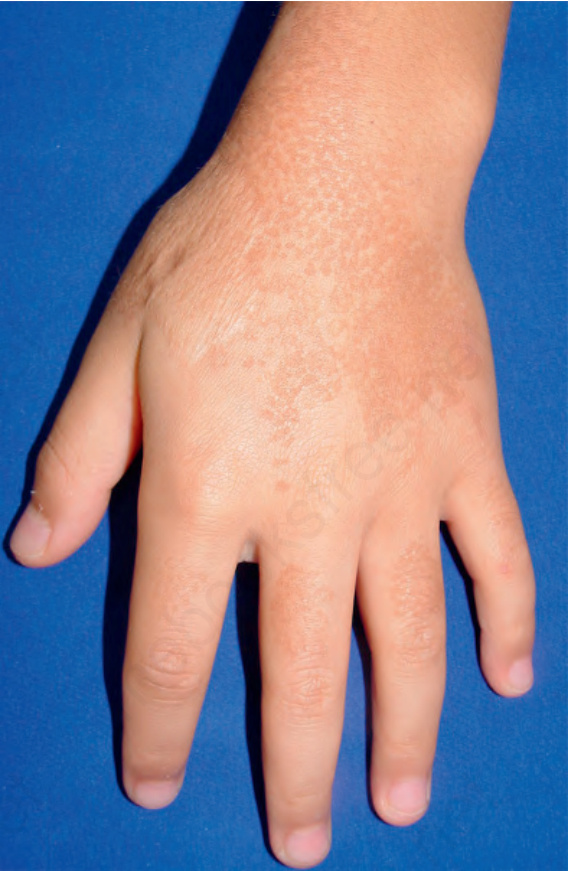
Fig. 20.31 Reticulate acropigmentation of Kitamura (macular reticulate pigmentation). By courtesy of Dr. Chao Sheau-Chiou, Tainan, Taiwan.