臨床特徵 (Clinical Features)
多發性肌炎 (polymyositis) 是一種罕見的肌肉發炎性疾病,病因不明。若同時出現某些特定的皮膚病灶,則使用「皮肌炎 (dermatomyositis)」一詞。整體發生率約為每年每百萬人口中有五例新住院病例。由於許多疾病都可能包含肌肉無力與肌肉酵素活性升高的特徵,因此診斷這兩種疾病時必須採用嚴格的標準 (Table 17.12)。若病人符合前四項標準(polymyositis),或符合四項中的三項再加上典型皮疹(dermatomyositis),即可確診其中任一狀況。
本病可分為五種變異型 (Table 17.13)。就第 V 型而言,dermatomyositis 最常與 scleroderma 共存(即 sclerodermatomyositis),但也可能與 SLE、rheumatoid arthritis 及 Sjögren syndrome 合併發生。要診斷為重疊症候群 (overlap syndrome),必須符合每一種疾病各自適當的診斷標準,而不能僅依據少數幾項共同表現。重疊症候群在 polymyositis 中比在 dermatomyositis 中更常發生,且呈現明顯的女性優勢(9:1)。
Dermatomyositis 並不罕見地僅以皮膚表現來呈現。此種表現型態的引述頻率有所差異。
對稱性近端肢體肌肉與頸前屈肌 (anterior neck flexors) 的無力,加上食道與呼吸肌受侵犯;肌肉切片陽性特徵;骨骼肌酵素升高;適當的肌電圖特徵;典型皮疹。
After Bohan, A. and Peter, J.B. (1975) Parts 1 and 2. New England Journal of Medicine, 292, 344–347, 403–407.
型別 變異型
I Polymyositis(多發性肌炎)
II Dermatomyositis(皮肌炎)
III 第 I 或 II 型併惡性腫瘤 (malignancy)
IV 兒童期 polymyositis 或 dermatomyositis
V 重疊症候群 (Overlap syndromes)
After Bohan, A. and Peter, J.B. (1975) Parts 1 and 2. New England Journal of Medicine, 292, 344–347, 403–407.
817 Polymyositis/dermatomyositis
其他皮膚特徵包括位於掌指關節 (metacarpophalangeal joints) 上方的紅斑性丘疹(Gottron sign)、甲周紅斑 (periungual erythema)、毛細血管擴張 (telangiectasia) 及線狀出血 (splinter hemorrhages)(Figs 17.128 and 17.129)。Gottron papules 典型出現於指關節,但膝、肘及踝(malleoli)也可能受影響。腳趾則具特徵性地不被侵犯。甲褶微血管 (nail fold capillaries) 可能腫大、擴張且扭曲變形。也常出現無血管區 (avascular areas)。有時可見甲小皮過度生長 (cuticular overgrowth),偶爾出現皮膚血管炎 (cutaneous vasculitis),表現為指端潰瘍 (digital ulceration)、甲周梗塞 (periungual infarcts) 及口腔潰瘍 (mouth ulcers),不過這在兒童期變異型中更常見。隨著時間進展,皮膚可能變得更萎縮並顯現多形性皮膚萎縮 (poikiloderma) 的特徵,特別影響伸側表面與上背部,但也可能範圍更廣(Fig. 17.130)。頭皮受侵犯並不少見,表現為脫屑與紅斑,且常伴搔癢。偶有光敏感 (photosensitivity) 的報告。其他罕見或不尋常的特徵包括牙齦毛細血管擴張 (gingival telangiectases)、類似 pityriasis rubra pilaris 的毛囊性丘疹(dermatomyositis 的 Wong 變異型)、紅皮症 (erythroderma)、侷限於脂漏部位的紅斑、伴特徵性風疹塊樣 (wheal-like) 表現的鞭笞狀紅斑 (flagellate erythema)、皮膚劃紋症 (dermographism)、類似惡性萎縮性丘疹病 (malignant atrophic papulosis,Degos disease) 的病灶、水疱大疱性皮疹 (vesiculobullous rash)、膿疱性疹 (pustular eruption)、類似 Sweet syndrome 的皮膚病、環狀肉芽腫 (granuloma annulare)、皮膚澱粉樣變性 (cutaneous amyloidosis)、局部黏液沉積症 (localized mucinosis)、脂膜炎 (panniculitis)、脂質失養症 (lipodystrophy),以及急性全身性
818 Idiopathic connective tissue disorders
皮下水腫 (subcutaneous edema)。牙齦毛細血管擴張也曾發現與 anti-Jo-1 antibody 相關。有些病人表現出影響軀幹與近端肢體的向心性鞭笞狀紅斑 (centripetal flagellate erythema)。可能發生表皮下水疱形成 (subepidermal blistering),且此現象已被連結到內臟惡性腫瘤。曾記錄一例罕見病例為急性發作的水疱大疱性病灶並合併腸道大量黏膜壞死。Bullous pemphigoid 也偶可罕見地與 dermatomyositis 相關。曾在一名以 prednisolone 與 methotrexate 治療的 dermatomyositis 病人身上報告爆發性皮膚纖維瘤 (eruptive dermatofibromas)。
對稱性近端(肢帶區,limb girdle)肌肉無力是 polymyositis 最常見的初發表現。雙腿幾乎總是最初受侵犯的部位。病人在從椅子起身、上樓梯、梳頭或從枕頭上抬頭時感到困難。有趣的是,顏面肌肉幾乎從不受侵犯。雖然肌肉可能會疼痛,但通常不嚴重,且壓痛也不常出現。肌肉萎縮在疾病病程後期才發展,此時可能併發纖維化與麻煩的攣縮 (contractures)。
食道受侵犯表現為吞嚥困難 (dysphagia),與合併惡性腫瘤的存在相關。症狀也可能因環咽橫紋肌 (cricopharyngeal striated muscle) 無力而顯示為食道前 (pre-esophageal) 受侵犯。後遺症包括鼻腔逆流 (nasal regurgitation) 與吸入性肺炎 (aspiration pneumonitis),
兒童期變異型具特徵性地出現皮膚血管炎 (cutaneous vasculitis),可能侵犯內臟;此情形也曾在成人病人中被描述,並可能與惡性腫瘤風險增加相關。指端潰瘍、甲周梗塞及口腔潰瘍是相關表現。在成人發病的 dermatomyositis 中,曾罕見報告與血管炎無關的深部皮膚與皮下潰瘍,這些可能與閉塞性(微)血管病變 (obliterative (micro)vasculopathy) 有關。皮膚、軟組織及肌肉的鈣化 (calcification) 罕見,但兒童期變異型例外,在該型中鈣化可能範圍廣泛並有助於診斷。皮膚鈣化已被證實與抗 140-kD 蛋白的自體抗體相關。
根據一項以台灣 2031 名病人為對象的大型族群研究指出,dermatomyositis/polymyositis 病人相較於配對對照組,靜脈血栓栓塞 (venous thromboembolism) 風險增加達 11 倍。
關節痛 (arthralgia) 並不少見,但明顯的關節炎 (arthritis) 則罕見,除非在重疊組的病人中。曾報告一名合併 anti-Jo-1 與抗環瓜胺酸胜肽抗體 (anticyclic citrullinated peptide antibodies) 的無肌病性皮肌炎 (amyopathic dermatomyositis) 病人出現破壞性關節病 (destructive arthropathy)。曾在單一病人報告鷹嘴突 (olecranon) 的無菌性滑囊炎 (aseptic bursitis)。
實驗室檢查可能顯示非特異性發現,如 ESR 上升、高γ球蛋白血症 (hypergammaglobulinemia),以及偽陽性的 Wassermann reaction。抗核因子 (antinuclear factor) 可在一小部分 polymyositis/dermatomyositis 病人中發現。Anti-RNP 與 -SM 抗體僅見於「重疊 (overlap)」病人。除了 anti-Jo-1 antibody 之外,其他新近描述的抗體還包括 PM-1、Ku、Mi-1、-2 及 -3,以及 Pa-1。這些抗體(除 anti-Jo-1 外)的意義尚不確定。
後者與高死亡率相關。聲音改變是一種不算少見的表現。幼年型皮肌炎 (Juvenile dermatomyositis) 也曾與缺血性潰瘍性結腸炎 (ischemic ulcerative colitis) 及乳糜瀉 (celiac disease) 相關。
Polymyositis/dermatomyositis 的肌電圖 (electromyographic) 特徵被認為具特徵性 (pathognomonic),包括以下三聯徵:靜止時的纖維顫動 (fibrillation at rest) 伴隨插入活動增加與正向尖波 (positive sharp waves)、短時程長振幅的多相電位 (polyphasic potentials),以及怪異的高頻重複放電 (bizarre high-frequency repetitive discharges)。
Polymyositis/dermatomyositis 常被強調的是與惡性腫瘤發生風險增加的關聯性。雖然小型研究所報告的發生率範圍極廣,從 15% 到 60% 的病例不等,但近期研究顯示此風險較低。惡性腫瘤的風險在 dermatomyositis 病人中(23%)似乎高於 polymyositis(8.9%)。Polymyositis/dermatomyositis 曾被描述與下列惡性腫瘤相關:乳癌 (breast cancer)、肺神經內分泌癌 (neuroendocrine carcinoma of the lung)、小細胞肺癌 (small cell lung cancer)、肝細胞癌 (hepatocellular carcinoma)、肝神經內分泌癌 (neuroendocrine carcinoma of the liver)、十二指腸類癌 (duodenal carcinoid)、膽囊癌 (gallbladder carcinoma)、膀胱癌 (carcinoma of the bladder)、攝護腺癌 (prostate cancer)、腎細胞癌 (renal cell carcinoma)、卵巢透明細胞癌 (clear cell ovarian carcinoma)、輸卵管癌 (fallopian tube cancer)、子宮癌肉瘤 (carcinosarcoma of the uterus)、鼻咽癌 (nasopharyngeal carcinoma)、食道癌 (esophageal cancer)、Klatskin tumor(肝門膽管癌,hilar cholangiocarcinoma)、甲狀腺癌 (thyroid cancer)、胸腺癌 (thymic carcinoma)、結腸癌 (cancer of the colon)、原發性胃黑色素瘤 (primary gastric melanoma)、轉移性黑色素瘤 (metastatic melanoma)、瀰漫性大 B 細胞淋巴瘤 (diffuse large B-cell lymphoma)、原發性皮膚 B 細胞淋巴瘤-腿型 (primary cutaneous B-cell lymphoma-leg type)、濾泡性淋巴瘤 (follicular lymphoma)、淋巴漿細胞樣淋巴瘤 (lymphoplasmocytoid lymphoma)、急性骨髓性白血病 (acute myeloid leukemia),以及 Kaposi sarcoma。在這些報告的關聯中,乳房、胃及卵巢腫瘤最常被引述。在一項近期分析中國 dermatomyositis 病人的研究中,鼻咽癌是最常見的關聯,其次為肺癌。病人應接受非常徹底的身體檢查,並結合常規實驗室檢查、胸部 X 光、腹部及骨盆腔的 CT 掃描,以及(女性病人的)乳房攝影 (mammography)。對於治療無反應或頻繁發生肌炎發作的病人,應懷疑潛在的惡性腫瘤。近期一項研究指出,需要更廣泛搜尋惡性腫瘤的病人包括:有全身性症狀 (constitutional symptoms) 者、dermatomyositis 或 polymyositis 快速發作者、無 Raynaud phenomenon 者、ESR 很高者,以及肌酸激酶 (creatine kinase) 值非常高者。
血清中通常含有升高的肌酸激酶 (creatine kinase)、醛縮酶 (aldolase)、乳酸脫氫酶 (lactate dehydrogenase) 及轉胺酶 (transaminases);由於任一病人不一定全部都會升高,因此通常建議常規檢測全部項目。連續性肌肉酵素研究對於監測病程進展與治療反應特別有用。
心肌受侵犯並不少見,病人可能出現心搏過速 (tachycardia)、竇性心搏過緩 (sinus bradycardia)、心電圖異常(例如束支傳導阻滯,bundle branch block)、充血性心衰竭 (congestive heart failure) 及心臟肥大 (cardiomegaly)。曾在一名 dermatomyositis 病人描述限制型心肌病變 (restrictive cardiomyopathy)。一項來自 British Columbia、納入 774 名發炎性肌肉病變 (inflammatory myopathies) 病人的大型族群研究顯示,此組病人心肌梗塞 (myocardial infarction) 風險顯著增加,尤其在診斷後第一年內。
肺部受侵犯(以間質纖維化 (interstitial fibrosis) 的影像學變化及/或呼吸功能受損的臨床證據判定)可能發生於多達 40% 的 polymyositis/dermatomyositis 病人。病人發生肺動脈高壓 (pulmonary hypertension) 的風險也增加。縱膈氣腫 (pneumomediastinum) 與間質性肺炎 (interstitial pneumonia) 是罕見併發症。肺含鐵血黃素沉著症 (pulmonary hemosiderosis) 是幼年型皮肌炎中極為例外的發現。近期一項重要的描述是 anti-Jo-1 antibody、肺纖維化 (pulmonary fibrosis) 與 dermatomyositis 三者之間的關聯。超過 50% 具有 anti-Jo-1 antibody 的病人有間質性肺病 (interstitial lung disease)。具此變異型的病人並無內臟惡性腫瘤發生率增加的風險。此變異型的其他特徵可能包括 Raynaud phenomenon、arthritis 及腱鞘炎 (tenosynovitis)。擴散至胸部肌肉可導致嚴重的呼吸困難;因此末期支氣管肺炎 (terminal bronchopneumonia) 是重要的死因。
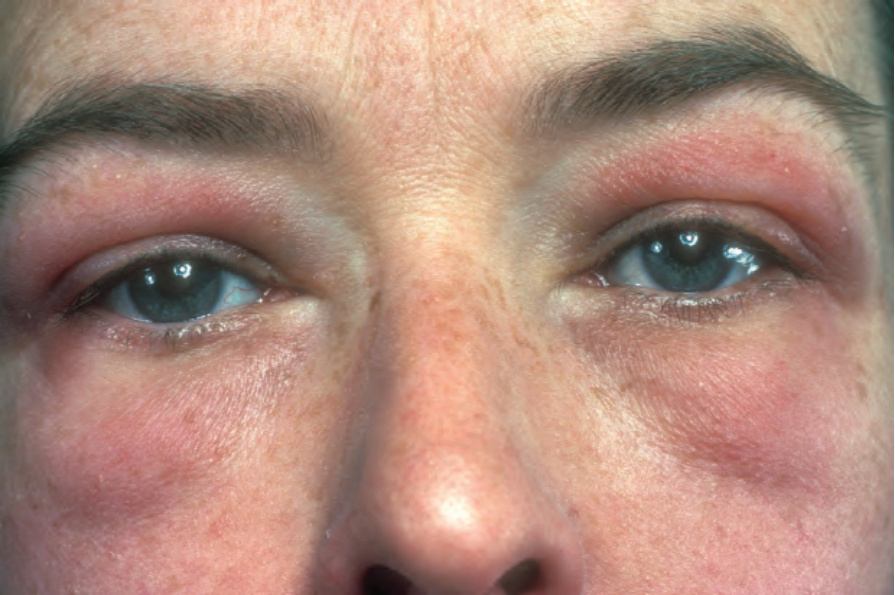
圖 17-125:皮肌炎 (dermatomyositis):注意眼周特徵性的紅紫色變色 (red-mauve discoloration)。也擴散至雙頰。From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
Fig. 17.125 Dermatomyositis: note the characteristic red-mauve discoloration around the eyes. There is also spread onto the cheeks. From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
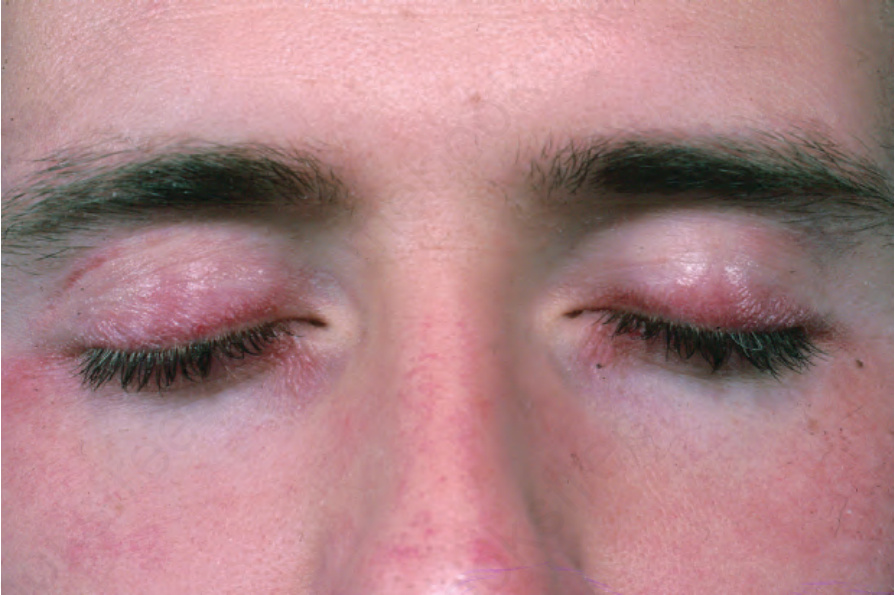
圖 17-126:皮肌炎 (dermatomyositis):上眼瞼特別受影響。From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
Fig. 17.126 Dermatomyositis: the upper eyelids are particularly affected. From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
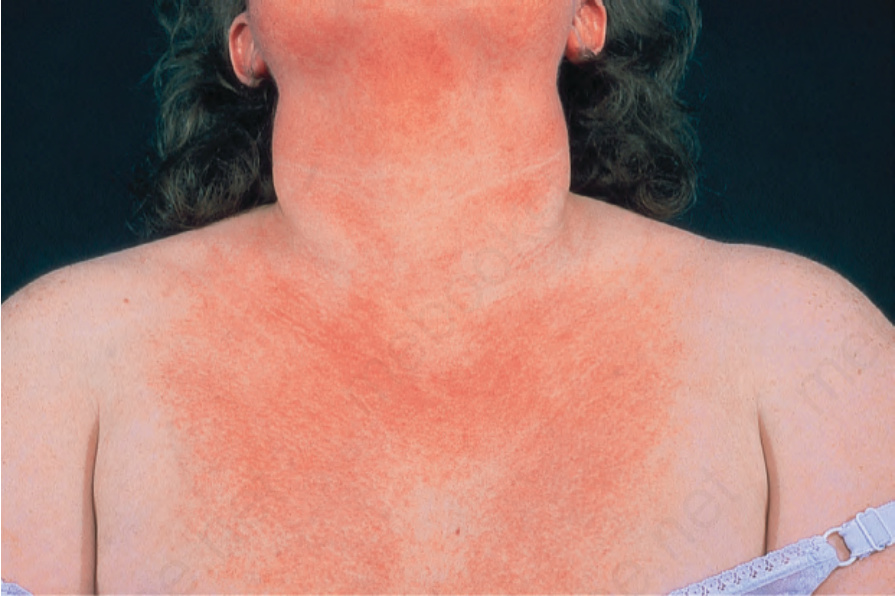
圖 17-127:皮肌炎 (dermatomyositis):注意此病人胸部的紅斑與輕微脫屑。By courtesy of the Institute of Dermatology, London, UK.
Fig. 17.127 Dermatomyositis: note the erythema and slight scale on this patient’s chest. By courtesy of the Institute of Dermatology, London, UK.
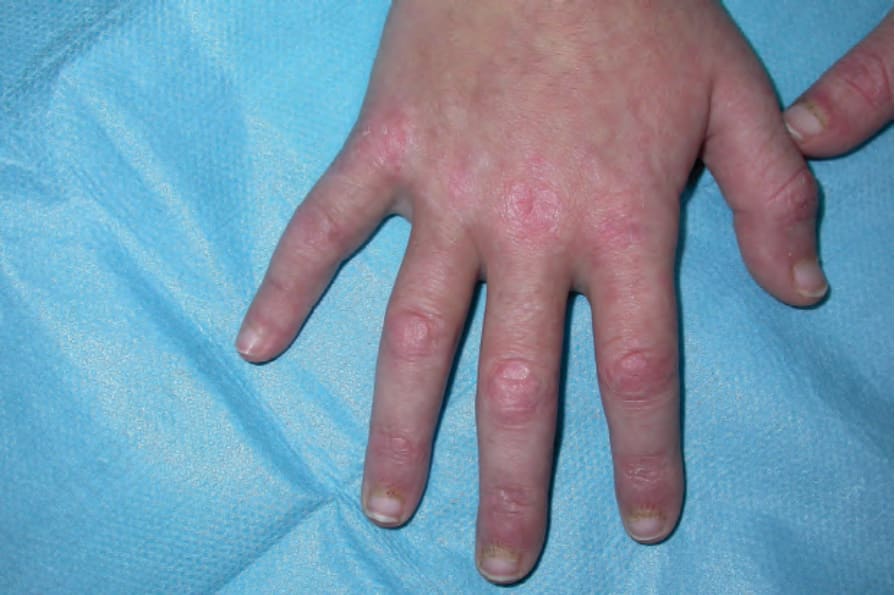
圖 17-128:皮肌炎 (dermatomyositis):指關節上特徵性的紫色丘疹(Gottron sign)。By courtesy of Dr J.C. Pascual, Alicante, Spain.
Fig. 17.128 Dermatomyositis: characteristic purple papules on the knuckles (Gottron sign). By courtesy of Dr J.C. Pascual, Alicante, Spain.
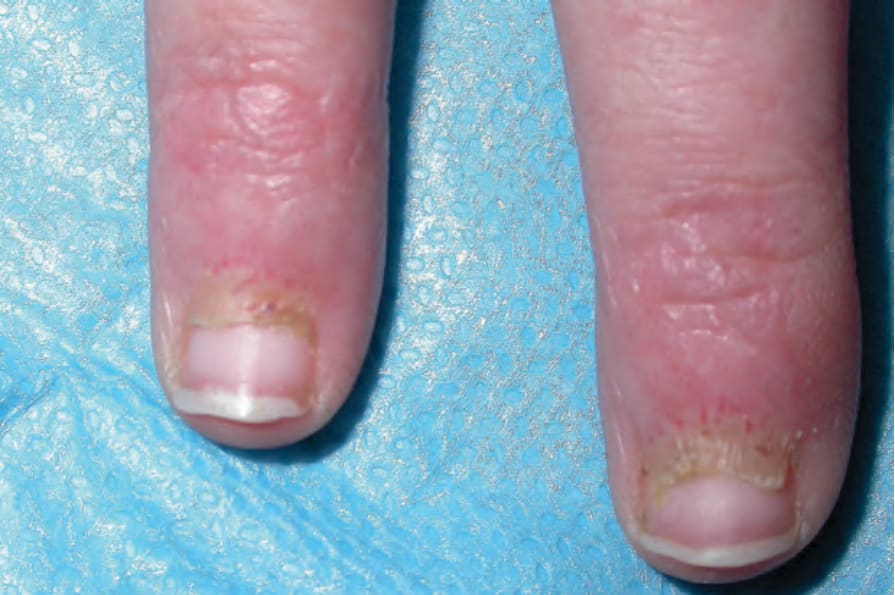
圖 17-129:皮肌炎 (dermatomyositis):Gottron papules、甲周紅斑 (periungual erythema) 及毛細血管擴張性的微血管環 (telangiectatic capillary loops)。By courtesy of Dr J.C. Pascual, Alicante, Spain.
Fig. 17.129 Dermatomyositis: Gottron papules, periungual erythema, and telangiectatic capillary loops. By courtesy of Dr J.C. Pascual, Alicante, Spain.
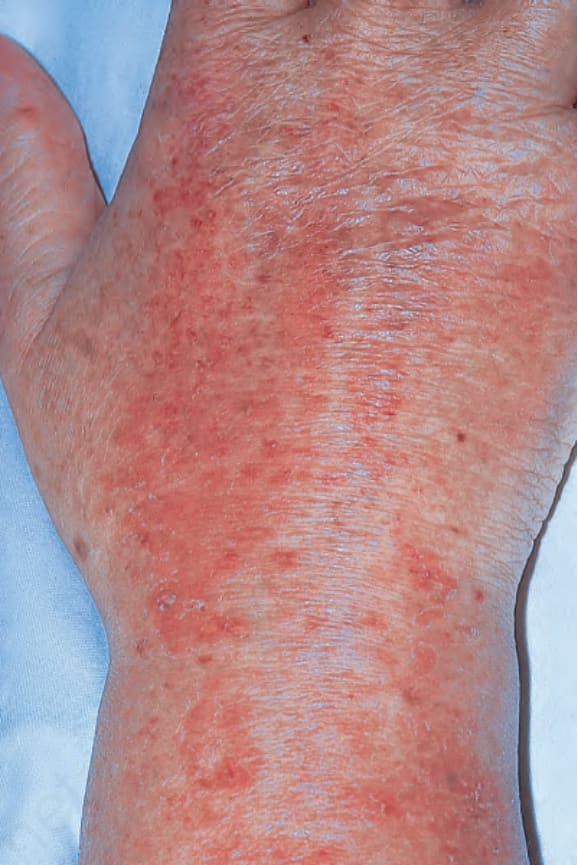
圖 17-130:皮肌炎 (dermatomyositis):此名長期罹病的病人,手背上出現萎縮與不定程度的色素變化(多形性皮膚萎縮,poikiloderma)。By courtesy of the Institute of Dermatology, London, UK.
Fig. 17.130 Dermatomyositis: in this patient with longstanding disease, atrophy and variable pigmentary changes (poikiloderma) are present on the dorsum of the hand. By courtesy of the Institute of Dermatology, London, UK.
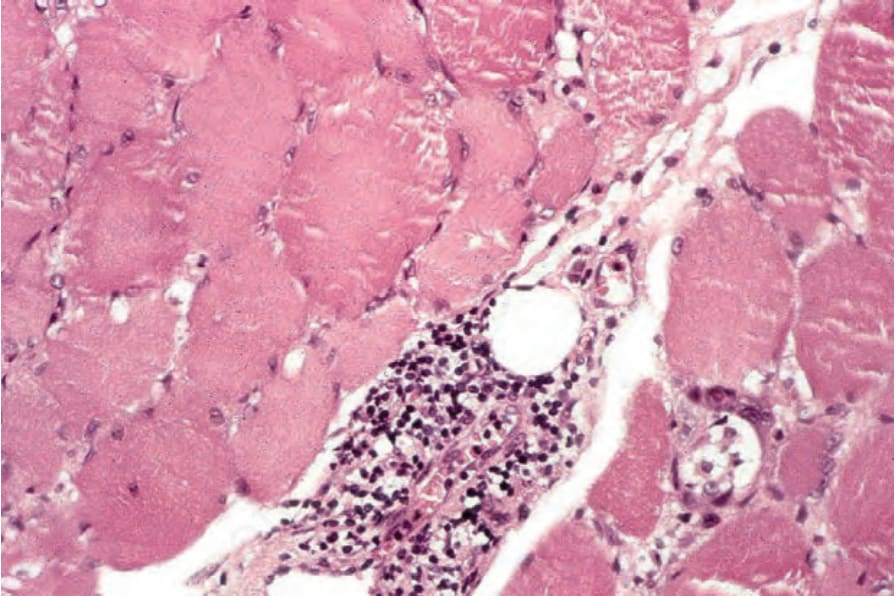
圖 17-136:皮肌炎 (dermatomyositis):浸潤主要由淋巴球 (lymphocytes) 組成。
Fig. 17.136 Dermatomyositis: the infiltrate consists predominantly of lymphocytes.

表 17-12:多發性肌炎/皮肌炎的診斷標準 (diagnostic criteria for polymyositis/dermatomyositis)。
Table 17.12 Diagnostic criteria for polymyositis/dermatomyositis
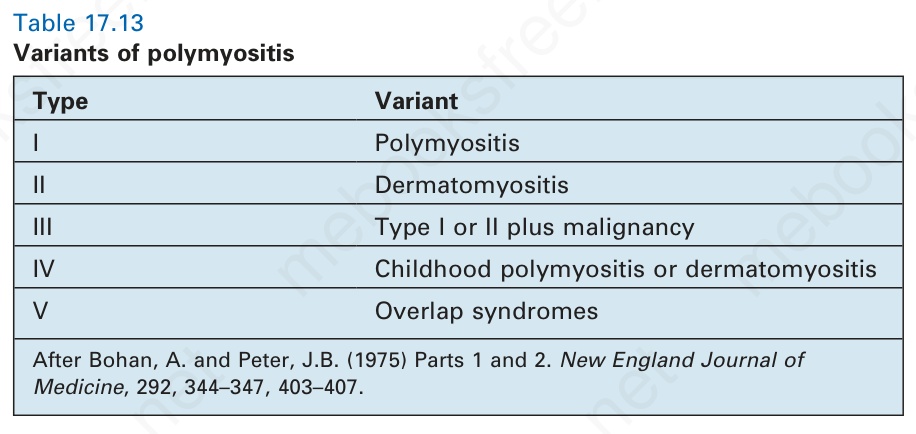
表 17-13:多發性肌炎的變異型 (variants of polymyositis)。
Table 17.13 Variants of polymyositis
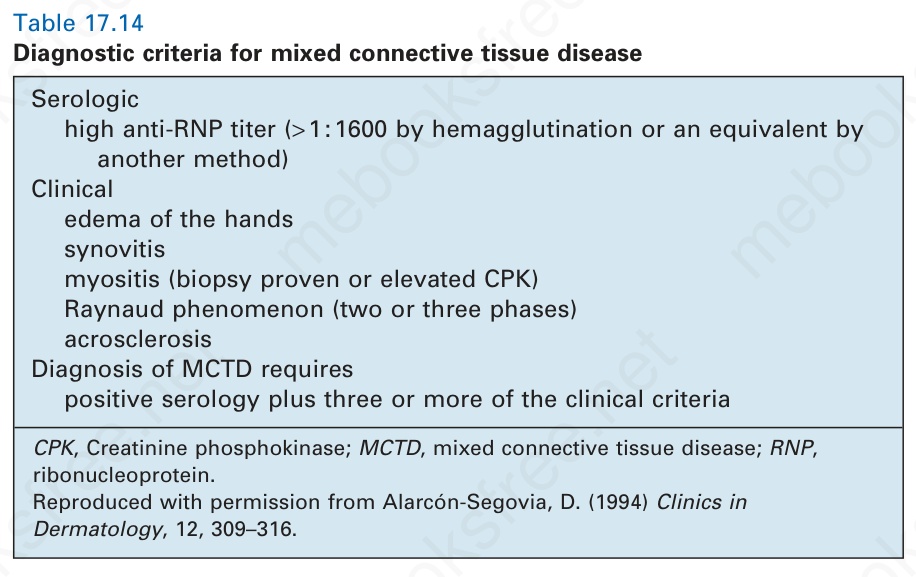
表 17-14:混合性結締組織病的診斷標準 (diagnostic criteria for mixed connective tissue disease)。
Table 17.14 Diagnostic criteria for mixed connective tissue disease
似乎有甲狀腺疾病風險增加的情形,特別是甲狀腺功能低下 (hypothyroidism),尤其在合併間質性肺病的病人中。Dermatomyositis 病人發生潰瘍性結腸炎 (ulcerative colitis) 的風險也增加。
幼年型皮肌炎 (Juvenile dermatomyositis) 的年發生率約為每年每百萬人口一例新病例,呈現女性優勢(2:1),最常於人生第一個十年發病。除了上述特徵外,還有高比例的血管病變性 (vasculopathic) 表現,包括可能致命的伴出血之胃腸道潰瘍 (gastrointestinal ulceration)。多器官受侵犯很常見。此病常以一次感染為前驅。預後通常良好,多達 70% 的兒童可完全康復。在嚴重受影響的病人中,
廣泛的皮膚侵犯可能因廣泛的疤痕形成與瀰漫性鈣化 (diffuse calcification) 而變得複雜。
硬皮病/多發性肌炎重疊 (Scleroderma/polymyositis overlap,即 sclerodermatomyositis) 是最常見的重疊症候群。雖然其肌炎成分通常與 dermatomyositis/polymyositis 所見者相同,但向陽性紅斑 (heliotrope erythema) 與 Gottron papules 通常不存在。硬皮病樣的皮膚表現傾向侷限於周邊部位。此重疊症候群與 Ku antibody 相關,並曾有一例報告與 Graves disease 及血小板減少性紫斑 (thrombocytopenic purpura) 相關。具有 anti-Ku antibody 的自體免疫性原發性血小板減少症 (Autoimmune idiopathic thrombocytopenia) 也曾與 dermatomyositis 相關。
819 Polymyositis/dermatomyositis
細胞媒介免疫 (Cell-mediated immunity) 在多發性肌炎實驗模型的發展中很重要。取自患有過敏性肌炎(基於連續注射異種肌肉加 Freund adjuvant)動物的淋巴球,在培養中對骨骼肌纖維具有細胞毒性,並可能發生淋巴母細胞轉化 (lymphoblastic transformation)。在人類疾病中確實存在類似情形,但這些是代表起始因子,還是作為肌肉損傷的後果而發展,則尚不清楚。已記錄各種細胞免疫異常,包括骨骼肌內出現活化的單核細胞、單核細胞向骨骼肌的異常遷移 (abnormal trafficking)、自體混合淋巴球反應 (autologous mixed lymphocyte responses) 降低,以及對自體肌肉的有絲分裂與增殖反應。
致病機轉與組織學特徵 (Pathogenesis / Histologic Features)
雖然 polymyositis/dermatomyositis 的病因與致病機轉不明,但有人提出環境因子(例如藥物、毒素或病毒)在與遺傳易感性 (genetic predisposition) 共同作用下,導致一種主要為免疫媒介 (immune-mediated) 的疾病。有證據顯示體液性 (humoral) 與細胞媒介 (cell-mediated) 成分皆很重要。
抗核因子 (antinuclear factor) 常存在。曾描述抗肌凝蛋白 (antimyosin) 與抗肌紅蛋白 (antimyoglobin) 抗體,但其意義不確定。目前不清楚這些抗體是先於還是隨著肌炎發作而出現,且其存在無法解釋皮膚表現。然而,抗肌凝蛋白抗體會伴隨任何發炎性肌炎出現,因此很可能是肌肉壞死的結果。
有些證據顯示存在遺傳性易感傾向,dermatomyositis 與 polymyositis 兩者中 HLA-B8 與 HLA-DR3 的發生率皆增加,特別是在具有 anti-Jo-1 antibodies 的病人中。家族性疾病的案例罕見。
許多動物實驗模型對人類肌炎可能的致病機轉提供了一些線索。將肌肉萃取物注射至多種動物體內,會導致一種輕度、非持續性的肌炎。
數種病毒——包括 Coxsackie B virus、猿猴後天免疫缺乏反轉錄病毒 (simian acquired immunodeficiency retrovirus) 及小鼠腦心肌炎病毒 (murine encephalomyocarditis virus)——已被證明可誘發一種慢性肌炎樣疾病。病毒株與宿主遺傳因子似乎特別重要。雖然不確定,但有人提出某些 dermatomyositis 病例,特別是幼年型變異型,可能代表對病毒感染的異常免疫反應。微小核糖核酸病毒 (Picornaviruses),包括 coxsackievirus 群,曾被特別牽涉其中。Anti-Jo-1 antibody(一種抗胺醯-tRNA 合成酶,antiaminoacyl-tRNA synthetase)會與組胺醯-轉移 RNA 合成酶 (histidyl-transfer RNA synthetase) 反應。此酵素已被證明除了其正常受質 tRNA 之外,還能與數種 picornaviruses 的 RNA 交互作用。有人提出此自體抗體的產生可能是這種異常交互作用的結果。
在 35% 至 40% 的 dermatomyositis/polymyositis 病人中,曾描述另一組針對核抗原 (nuclear antigens) 的抗體:
• PM-1 (PM-Scl) antibody 與 polymyositis 及 polymyositis/scleroderma 重疊密切相關。
• Ku antibody 是 sclerodermatomyositis 的標記。
• PA-1 antibody 與 polymyositis、arthritis 及纖維化性肺泡炎 (fibrosing alveolitis) 相關。
• Mi-2 與 dermatomyositis 相關。
對 RNP 抗原 U1 與 U2 的抗體存在,雖然不具特異性,但確實高度提示 dermatomyositis/systemic sclerosis 重疊症候群。抗訊號辨識顆粒抗體 (Antisignal recognition particle (SRP) antibodies) 並不常見,但通常與嚴重疾病相關。雖然這些抗核自體抗體具診斷價值,但尚未被證明具有致病學上的意義。
值得注意的是,一種類似 dermatomyositis 的疾病可由數種感染性病原體誘發,包括 leishmania、parvovirus(erythrovirus)B19、人類免疫缺乏病毒 (human immunodeficiency virus) 及 toxoplasma。曾有一名 dermatomyositis 病人發生結核性肌筋膜炎 (tuberculous myofasciitis)。Dermatomyositis/polymyositis 曾被報告為多種藥物的不良反應,例如 hydroxyurea、cyclophosphamide、etoposide、fluvastatin、simvastatin、pravastatin、atorvastatin、ipilimumab、capecitabine、anti-TNF alpha 治療、omeprazole、minocycline、carbimazole、terbinafine 及 interferon beta-1a。此外,polymyositis/dermatomyositis 也曾發現與 B 型肝炎疫苗接種 (hepatitis B vaccination)、psoriasis、pemphigus foliaceus、Duchenne muscular dystrophy 帶因狀態、家族性大腸瘜肉症 (familial polyposis colli)、ulcerative colitis、噬血症候群 (hemophagocytic syndrome)、有機溶劑 (organic solvent),以及矽膠填充式乳房植入物 (silicone gel-filled breast implants) 相關。
近期已鑑定出數種肌炎特異性抗體 (myositis-specific antibodies),與疾病表型密切相關。抗轉錄中間因子 1 抗體(anti-transcriptional intermediary factor 1,anti-TIF1;先前命名為 anti-p155-kD protein antibodies)對成人與兒童的 dermatomyositis 特別具特異性,並在約 30% 的兩組病人中被偵測到。此外,anti-TIF1 antibodies 與惡性腫瘤強烈相關,特別是在 40 歲以上的 dermatomyositis 病人中。據報告,多達 75% 的成人 dermatomyositis 病人發展出惡性腫瘤。抗黑色素瘤分化抗原 5 抗體(anti-melanoma differentiation antigen 5,anti-MDA5;也稱為 anti-CADM-140 antibodies)在無肌病性皮肌炎 (amyopathic dermatomyositis) 病人中較常見,並在日本病人中與間質性肺病相關。此外,已證明 anti-MDA5 antibody 的濃度與疾病活動性相關,且疾病復發與 anti-MDA5 濃度的再次上升相關。這些數據顯示 anti-MDA5 antibody 濃度可用於監測疾病活動性,並作為無肌病性皮肌炎病人復發的預測標記。抗核基質蛋白 2 抗體(Antinuclear matrix protein 2 antibodies;原命名為 anti-MJ antibodies)在多達 25% 的幼年型肌炎病人中被偵測到,並與以肌肉攣縮 (muscle contractures) 與萎縮為特徵的嚴重疾病病程相關。此外,anti-NXP2 自體抗體的存在會顯著增加皮膚鈣質沉著症 (calcinosis cutis) 的風險。抗小泛素樣修飾子活化酵素抗體 (Anti-small ubiquitin-like modifier activating enzyme antibodies) 存在於少於 10% 的成人 dermatomyositis 病人中,並與高比例的嚴重吞嚥困難 (severe dysphagia) 相關。
數項研究已證實,相較於健康對照組,dermatomyositis/polymyositis 病人的帶狀疱疹 (herpes zoster) 發生率增加,尤其是在 50 歲以上、有一項或多項共病的女性中,這些共病包括糖尿病、腎臟疾病、肥胖、癌症、其他自體免疫疾病、MCTDs、血管炎,及/或接受免疫抑制藥物或皮質類固醇 (corticosteroids) 治療。
Dermatomyositis 與 polymyositis 可能發生於已知患有其他自體免疫疾病的病人,包括自體免疫性甲狀腺疾病 (autoimmune thyroid disease) 與胰島素依賴型糖尿病 (insulin-dependent diabetes mellitus)。體液免疫在 dermatomyositis 中的確切角色尚不清楚,但一般認為與微血管喪失 (capillary loss) 及缺血性損傷 (ischemic damage) 特別相關。
病灶皮膚的直接免疫螢光 (Direct immunofluorescence) 顯示,約 35% 的病人在真皮-表皮交界 (dermal–epidermal junction) 處有免疫球蛋白(IgG、IgA 及 IgM)與補體的顆粒狀沉積。部位選擇很重要,以甲床切片 (nail bed biopsies) 的陽性率最高。一項較近期的研究證明,C5b–9 沉積於血管壁與沿真皮-表皮交界處,且同時 lupus band test 為陰性。此發現具有高特異性(93.5%)與敏感性(78.5%)。表皮角質細胞 (epidermal keratinocytes) 也可能對 C5b–9 與 IgG 呈陽性。在小血管壁中發現 C5b–9 顯示,補體媒介的微血管損傷 (complement-mediated microvascular injury) 在 dermatomyositis 的致病機轉中可能有某種重要性。
兒童期 dermatomyositis 的致病機轉主要以缺血 (ischemic) 為基礎(見下文)。
皮膚發現各不相同。紅斑性疹顯示輕微的過度角化 (hyperkeratosis) 與表皮萎縮 (epidermal atrophy),並伴有表皮突型態的消失 (effacement of the ridge pattern)(Fig. 17.131)。基底細胞液化變性 (Basal cell liquefactive degeneration) 是典型表現,且
820 Idiopathic connective tissue disorders
有時可見類膠樣小體 (cytoid bodies)(Fig. 17.132)。基底膜增厚 (Basement membrane thickening) 偶爾很顯著。有上層真皮水腫 (upper dermal edema),且可能可見噬黑色素細胞 (melanophages)。罕見情況下,水腫會導致表皮下水疱形成 (subepidermal vesiculation)。通常存在輕度慢性發炎細胞浸潤 (light chronic inflammatory cell infiltrate)(Fig. 17.133)。其通常侷限於淺層真皮,且不與皮膚附屬器 (cutaneous adnexae) 相關。浸潤由活化的 T 淋巴球與巨噬細胞組成,偶有真皮 Langerhans 細胞。輔助 T 細胞 (Helper T cells) 占多數。在某些情況下,明顯的過度角化、毛囊角栓 (follicular plugging)、真皮水腫,以及增加的基底膜樣物質 (basement membrane-like material),會導致與紅斑性狼瘡 (lupus erythematosus) 有相當程度的組織學重疊,因此臨床病理對照 (clinicopathological correlation) 至關重要。近期一項研究發現,dermatomyositis 中最一致的組織學參數為基底角質細胞的空泡變化 (vacuolar changes of basal keratinocytes)、真皮黏液堆積 (dermal mucin accumulation),以及輕度至中度的真皮單核發炎細胞浸潤。真皮硬化 (Dermal sclerosis) 偶爾可能存在。
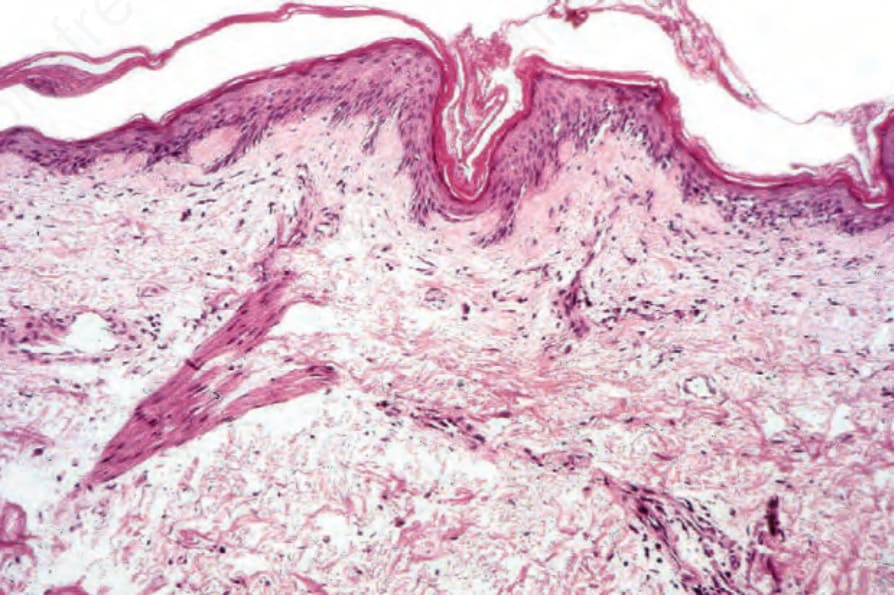
圖 17-131:皮肌炎 (dermatomyositis):有過度角化 (hyperkeratosis) 與表皮萎縮 (epidermal atrophy)。注意輕微的毛細血管擴張 (telangiectasia)。
Fig. 17.131 Dermatomyositis: there is hyperkeratosis and epidermal atrophy. Note the mild telangiectasia.
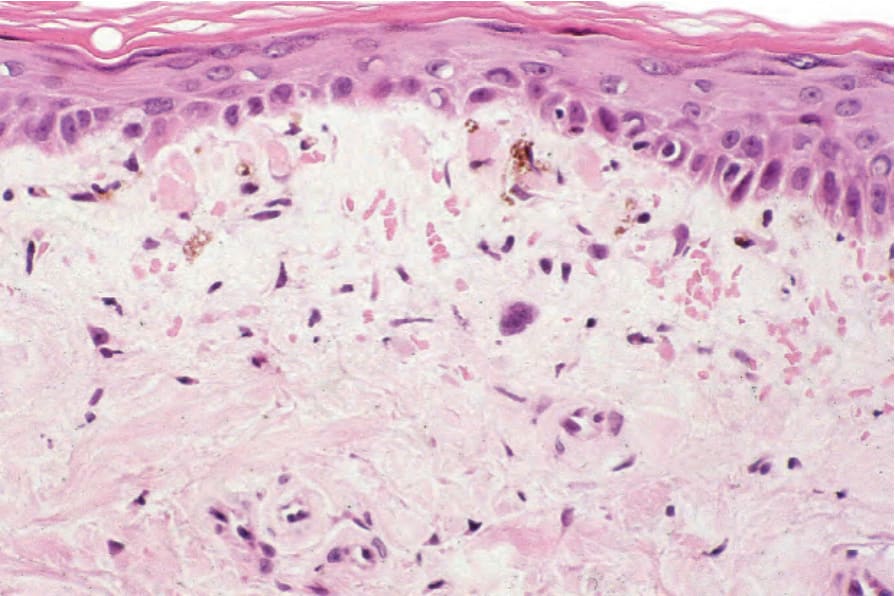
圖 17-132:皮肌炎 (dermatomyositis):有萎縮伴表皮突型態消失 (effacement of the ridge pattern)。在此例中類膠樣小體 (cytoid bodies) 很明顯。注意色素失禁 (pigmentary incontinence)。
Fig. 17.132 Dermatomyositis: there is atrophy with effacement of the ridge pattern. In this example cytoid bodies are conspicuous. Note the pigmentary incontinence.
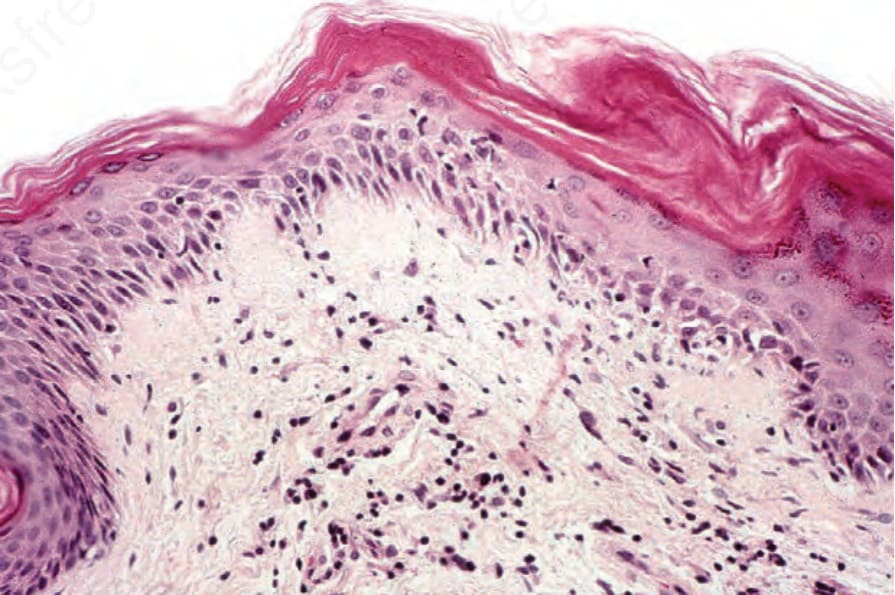
圖 17-133:皮肌炎 (dermatomyositis):右側可見局部、輕度的基底細胞水腫性變性 (basal cell hydropic degeneration)。存在慢性發炎細胞浸潤。
Fig. 17.133 Dermatomyositis: focal, mild basal cell hydropic degeneration is seen on the right. A chronic inflammatory cell infiltrate is present.
色素失禁 (incontinence)、類膠樣小體形成 (cytoid body formation),以及斑塊狀淋巴球性發炎細胞浸潤。真皮呈水腫狀,常含有增加的黏液 (mucin),並具特徵性地顯示明顯擴張的血管腔 (dilated vascular channels)。如多形性皮膚萎縮型蕈狀肉芽腫 (poikilodermatous mycosis fungoides) 中所見的浸潤細胞核非典型性 (nuclear atypia),並非本病特徵。
真皮內常存在增加量的 Alcian blue 陽性醣胺聚醣 (Alcian blue-positive glycosaminoglycans)。有時有鈣化灶 (foci of calcification),且偶爾可見脂膜炎 (panniculitis)。
多形性皮膚萎縮型病灶顯示過度角化、輕度表皮萎縮伴表皮突型態喪失,以及基底細胞液化變性。其他特徵可能包括明顯的色素
Gottron papules 的特徵為過度角化、輕度乳頭瘤狀增生 (mild papillomatosis)、棘層肥厚 (acanthosis),或較少見的表皮萎縮,以及如上所述的介面性皮膚炎 (interface dermatitis) 特徵(Fig. 17.134)。向心性鞭笞狀紅斑 (centripetal flagellate erythema) 的組織學顯示介面性皮膚炎的變化。
超微結構研究 (Ultrastructural studies) 對我們理解 dermatomyositis 貢獻甚少。如 SLE 中所描述的管網狀包涵體 (Tubuloreticular inclusions),曾在內皮細胞與周細胞 (pericyte) 的細胞質中被記錄,但其意義不確定。
在幼年型變異型中,皮膚特徵與上述者相似,但有以下但書:有時可見纖維化、鈣化較常見,且閉塞性血管疾病(以伴纖維蛋白血栓的纖維性內膜增生 (fibrous intimal proliferation with fibrin thrombi) 為特徵)常存在。
骨骼肌變化除了局部慢性發炎細胞浸潤外,還包括退化性 (degenerative) 與再生性 (regenerative) 特徵(Figs 17.135 and 17.136)。後者主要由淋巴球組成,但
821 Polymyositis/dermatomyositis
組織球 (histiocytes)、嗜酸性球 (eosinophils) 及漿細胞 (plasma cells) 也可能可見。淋巴球除了 T 細胞之外,還包含為數可觀的 B 細胞,特別是在與血管相關處;而 T 細胞則主要見於變化的肌纖維內及其周圍。如同皮膚病灶,輔助 T 細胞占多數。然而,多達 25% 的肌肉切片可能不顯示任何發炎證據。退化性纖維腫脹且呈強烈嗜酸性 (intensely eosinophilic),並可能顯示橫紋喪失 (loss of striations)(Fig. 17.137)。有些纖維呈空泡化,但其他則呈顆粒狀或碎裂狀 (granular or fragmented)(Fig. 17.138)。肌肉細胞核的增生與中央化 (centralization) 很常見,肌漿嗜鹼性 (sarcoplasmic basophilia) 亦然——這些都是再生的特徵(Fig. 17.139)。近期在 dermatomyositis 病人中也曾報告與包涵體肌炎 (inclusion body myositis) 完全相同的組織學變化,即鑲邊空泡 (rimmed vacuoles)。
若切片取自長期「燃盡 (burned out)」的病灶,則肌纖維呈萎縮,並有肌內膜纖維化 (endomysial fibrosis)。束周萎縮 (Perifascicular atrophy)——即在一個肌束邊緣出現一兩排萎縮纖維——被認為是 dermatomyositis 的特徵。據說 dermatomyositis 與 polymyositis 的肌肉病理有所不同。在 dermatomyositis 中,發炎細胞浸潤傾向於分布於間隔 (septal) 或血管周圍 (perivascular);而在 polymyositis 中則為肌束內 (intrafascicular)。Dermatomyositis 中的肌肉壞死傾向侵犯小群纖維,而在 polymyositis 中受影響的纖維則傾向於單一且稀疏。
去神經性神經病變特徵 (Denervation neuropathic features) 偶爾也存在,推測是由於發炎過程侵犯小的肌肉內
822 Idiopathic connective tissue disorders
神經纖維所致。在接受治療的病人切片中,可見第 II 型肌纖維的類固醇萎縮 (Steroid atrophy of type II muscle fibers)。
在兒童期 dermatomyositis 中,影響微血管、小靜脈 (venules) 及小動脈 (arterioles) 的血管變化很常見。發炎成分通常相當稀疏,由淋巴球、單核球 (monocytes) 及漿細胞組成,主要集中於束周結締組織 (perifascicular connective tissue) 內的血管系統。肌肉變化各不相同,從較輕微疾病中的束周萎縮,到較嚴重受影響病人中的局部壞死與梗塞,後者也可能以纖維化為特徵。血管病灶包括內皮細胞腫脹與壞死(伴或不伴閉塞)、非壞死性淋巴球性血管炎 (non-necrotizing lymphocytic vasculitis),以及周邊肌束微血管床 (peripheral fascicular capillary bed) 的喪失。
血清學 (Serologic)
高 anti-RNP 效價(以血球凝集法 (hemagglutination) 測得 > 1:1600,或以其他方法測得之等值)
臨床 (Clinical)
手部水腫 (edema of the hands);滑膜炎 (synovitis);肌炎(myositis,經切片證實或 CPK 升高);Raynaud phenomenon(兩相或三相);肢端硬化 (acrosclerosis)。MCTD 的診斷需要
兒童期 dermatomyositis 肌肉切片的免疫螢光 (Immunofluorescence) 常顯示血管壁內 (vascular intramural) 的 IgM 與 C3。
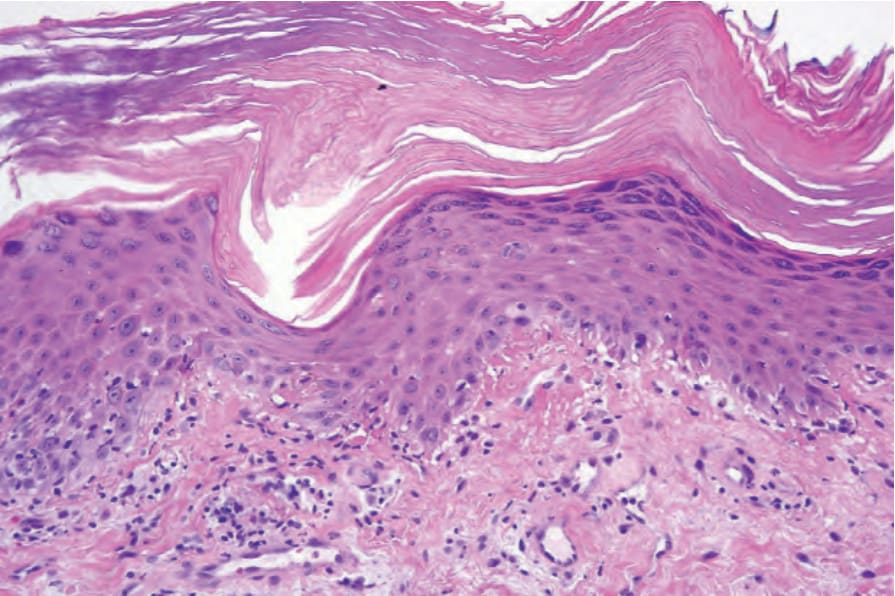
圖 17-134:Gottron papule:注意過度角化 (hyperkeratosis)、顆粒層增厚 (hypergranulosis) 及模擬扁平苔癬 (lichen planus) 的不規則棘層肥厚 (irregular acanthosis)。有基底細胞水腫性變性 (basal cell hydropic degeneration),且淺層真皮中存在類膠樣小體 (cytoid bodies)。By courtesy of D. Whittemore, DO, MD Anderson Cancer Center, Houston, Texas, USA.
Fig. 17.134 Gottron papule: note the hyperkeratosis, hypergranulosis, and irregular acanthosis simulating lichen planus. There is basal cell hydropic degeneration and cytoid bodies are present in the superficial dermis. By courtesy of D. Whittemore, DO, MD Anderson Cancer Center, Houston, Texas, USA.
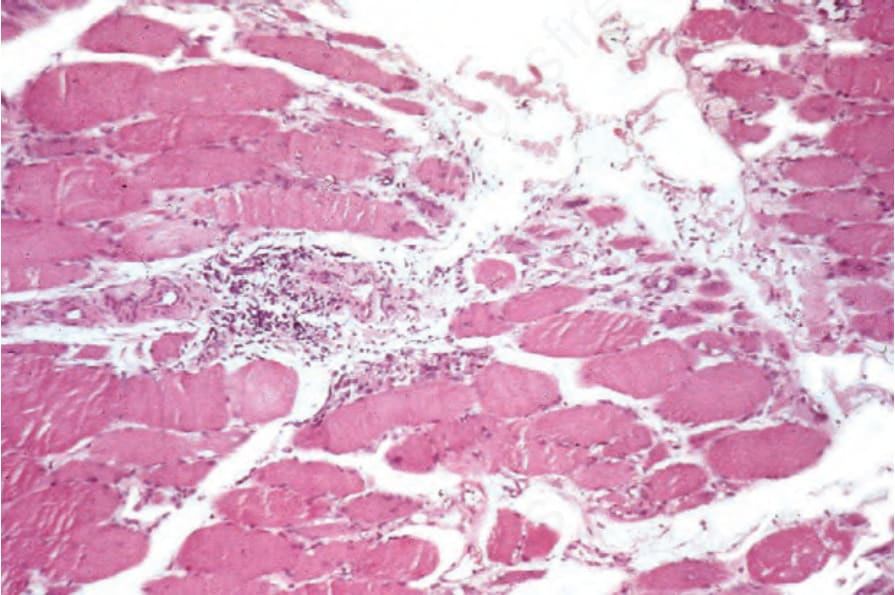
圖 17-135:皮肌炎 (dermatomyositis):注意血管周圍的慢性發炎細胞浸潤 (perivascular chronic inflammatory cell infiltrate)。
Fig. 17.135 Dermatomyositis: note the perivascular chronic inflammatory cell infiltrate.
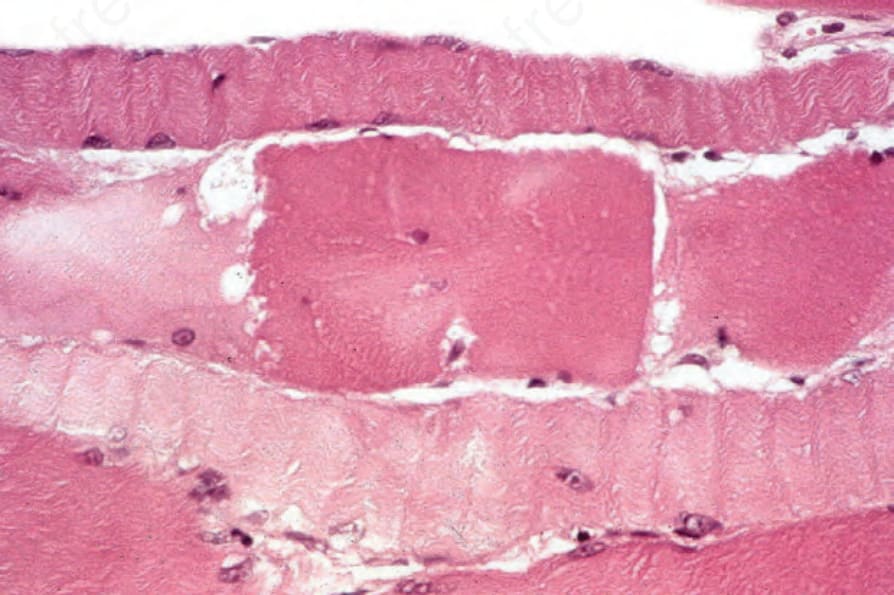
圖 17-137:皮肌炎 (dermatomyositis):中央的纖維強烈腫脹、嗜酸性且碎裂;有橫紋喪失 (loss of striations)。
Fig. 17.137 Dermatomyositis: the central fiber is intensely swollen, eosinophilic, and fragmented; there is a loss of striations.
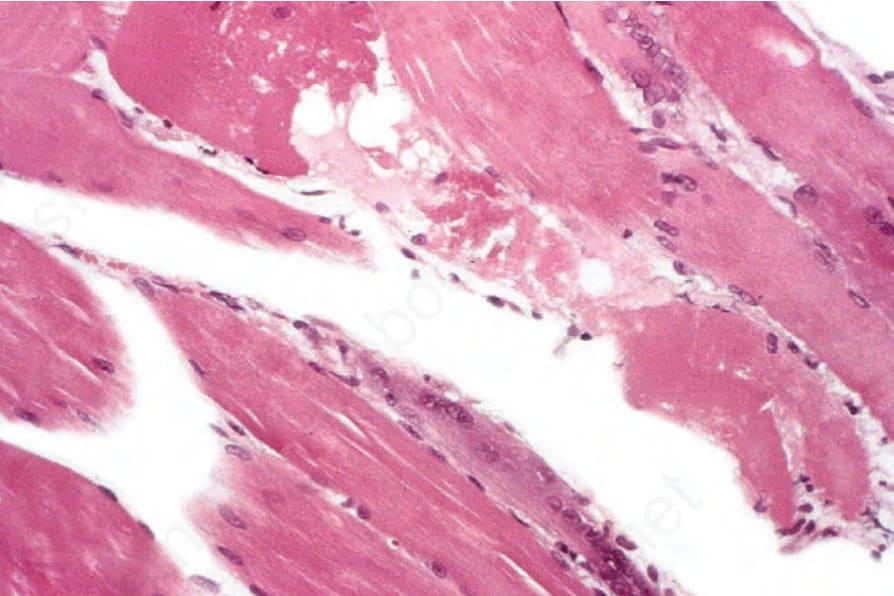
圖 17-138:皮肌炎 (dermatomyositis):上方中央區的纖維腫脹、嗜酸性、空泡化,且部分區域呈顆粒狀;其下方為一個再生中的嗜鹼性細胞 (regenerating basophilic cell)。
Fig. 17.138 Dermatomyositis: the fiber in the upper midfield is swollen, eosinophilic, vacuolated, and in places granular; beneath is a regenerating basophilic cell.
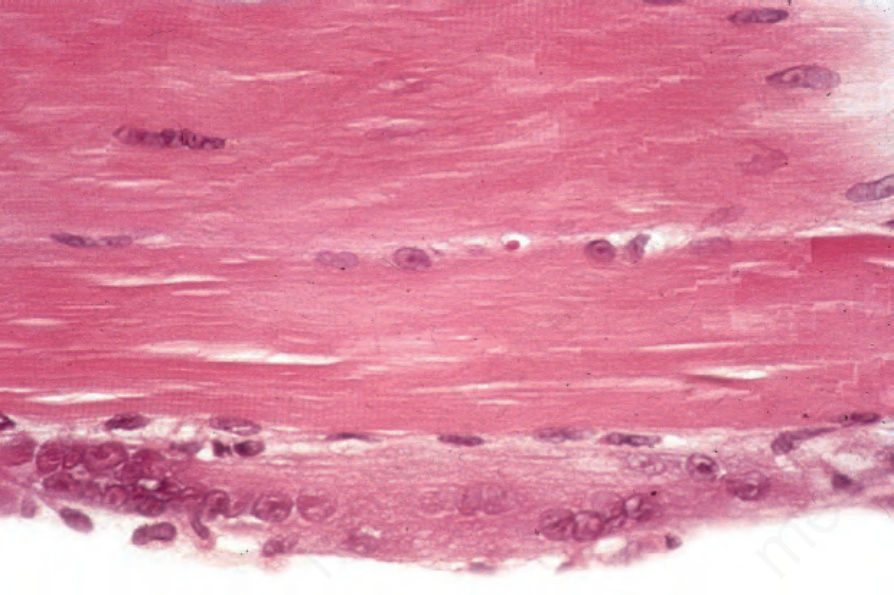
圖 17-139:皮肌炎 (dermatomyositis):下方纖維呈嗜鹼性並顯示過多的細胞核——再生的特徵。注意上方纖維中細胞核的中央化 (centralization of nuclei)。
Fig. 17.139 Dermatomyositis: the lower fiber is basophilic and shows excessive nuclei – features of regeneration. Note the centralization of nuclei in the upper fiber.