Polymyositis/dermatomyositis
Polymyositis/dermatomyositis
Clinical features Polymyositis is a rare inflammatory disorder of muscle, the etiology of which is unknown.1,2 If certain cutaneous lesions are also present, the term ‘dermatomyositis’ is applied. The overall incidence is approximately five new hospital cases per million of the population per year.1 As many diseases may include features of muscle weakness and elevated muscle enzyme activities, strict criteria must be applied to the diagnosis of these two diseases (Table 17.12). Either of these conditions may be confidently diagnosed if a patient fulfills the first four criteria (polymyositis) or three of the four plus the typical rash (dermatomyositis).3,4
Five variants of the disease are recognized (Table 17.13). With respect to type V, dermatomyositis most commonly coexists with scleroderma (sclerodermatomyositis), but it may also develop in association with SLE, rheumatoid arthritis, and Sjögren syndrome.4,5 To diagnose an overlap syndrome the appropriate diagnostic criteria must be fulfilled for each disease and not just a few common manifestations. Overlap syndromes occur more frequently in polymyositis than in dermatomyositis and show a marked female predominance (9 : 1).6,7
Dermatomyositis not uncommonly presents solely with cutaneous manifestations. The quoted frequency of this type of presentation is variable
Symmetrical weakness of proximal limb muscles and anterior neck
flexors, plus esophageal and respiratory muscle involvement Positive muscle biopsy features Elevated skeletal muscle enzymes Appropriate electromyographic features A typical rash
After Bohan, A. and Peter, J.B. (1975) Parts 1 and 2. New England Journal of Medicine, 292, 344–347, 403–407.
Type Variant
I Polymyositis
II Dermatomyositis
III Type I or II plus malignancy
IV Childhood polymyositis or dermatomyositis
V Overlap syndromes
After Bohan, A. and Peter, J.B. (1975) Parts 1 and 2. New England Journal of Medicine, 292, 344–347, 403–407.
817 Polymyositis/dermatomyositis
Other cutaneous features include erythematous papules over the metacarpophalangeal joints (Gottron sign), periungual erythema, telangiectasia, and splinter hemorrhages (Figs 17.128 and 17.129). Gottron papules are typically found on the knuckles, but knees, elbows, and malleoli may also be affected.22 The toes are characteristically spared. The nail fold capillaries may be enlarged, dilated, and distorted. Avascular areas are also often present.18 Cuticular overgrowth is sometimes evident, and occasionally there is a cutaneous vasculitis presenting as digital ulceration, periungual infarcts, and mouth ulcers, though more often in the childhood variant.18 With time, the skin may become more atrophic and show the features of poikiloderma, which particularly affects the extensor surfaces and upper back, but may be more widespread (Fig. 17.130).6,7 Scalp involvement, which presents with scaling and erythema, is not uncommon and is frequently pruritic.18 Photosensitivity has occasionally been reported.6,19 Other rare or unusual features include gingival telangiectases, follicular papules resembling pityriasis rubra pilaris (Wong variant of dermatomyositis), erythroderma, erythema confined to seborrheic areas, flagellate erythema with characteristic wheal-like manifestations, dermographism, lesions resembling malignant atrophic papulosis (Degos disease), a vesiculobullous rash, a pustular eruption, Sweet syndrome-like dermatosis, granuloma annulare, cutaneous amyloidosis, localized mucinosis, panniculitis, lipodystrophy, and acute generalized
subcutaneous edema.22–49 Gingival telangiectases have also been found in association with anti-Jo-1 antibody.50 Some patients present with a centripetal flagellate erythema affecting the trunk and proximal extremities.51 Subepidermal blistering may occur and this has been linked to internal malignancy.27,52,53 A rare case with acute onset vesiculobullous lesions and massive mucosal necrosis of the intestine has been documented.54 Bullous pemphigoid may also rarely be associated with dermatomyositis.55 Eruptive dermatofibromas have been reported in a single patient with dermatomyositis who was treated with prednisolone and methotrexate.56
Symmetrical proximal (limb girdle) muscle weakness is the most common presenting feature of polymyositis.6 The legs are almost always the initial site of involvement. The patient experiences difficulty in getting out of a chair, walking up the stairs, combing his hair, or raising his head from a pillow. Interestingly, the facial muscles are almost never involved.57 Although the muscles may be painful, this is not usually severe, and tenderness is not often present. Muscle atrophy develops later in the course of the disease when fibrosis and troublesome contractures may supervene.
Esophageal involvement manifests as dysphagia, which correlates with the presence of an associated malignancy.4 Symptoms may also indicate pre-esophageal involvement due to cricopharyngeal striated muscle weakness.6 Sequelae include nasal regurgitation and aspiration pneumonitis, the
818 Idiopathic connective tissue disorders
Cutaneous vasculitis is characteristic of the childhood variant, which may involve the viscera; it has also been described in adult patients and may be associated with an increased risk of malignancy. Digital ulcers, periungual infarcts, and oral ulcers are associated manifestations.6 Deep cutaneous and subcutaneous ulcers not associated with vasculitis, have rarely been reported in adult-onset dermatomyositis, and are likely to be related to obliterative (micro)vasculopathy.76 Calcification of the skin, soft tissues, and muscle is rare except in the childhood variant where it may be widespread and of help diagnostically.77–79 Skin calcification has been demonstrated to correlate with autoantibodies to a 140-kD protein.79
Patients with dermatomyositis/polymyositis exhibit 11-fold increased risk for venous thromboembolism in comparison with a matched control group, as suggested by a large population-based study on 2031 patients from Taiwan.80
Arthralgia is not uncommon, but frank arthritis is rare except in the overlap group of patients.4 Destructive arthropathy has been reported in a patient with amyopathic dermatomyositis associated with anti-Jo-1 and anticyclic citrullinated peptide antibodies.81 Aseptic bursitis of the olecranon has been reported in a single patient.82
Laboratory investigations may reveal non-specific findings of a raised ESR, hypergammaglobulinemia, and a false-positive Wassermann reaction. Antinuclear factor may be found in a small percentage of patients with polymyositis/dermatomyositis. Anti-RNP and -SM antibodies are only seen in ‘overlap’ patients. In addition to anti-Jo-1 antibody, additional newly described antibodies include PM-1, Ku, Mi-1, -2 and -3, and Pa-1.83 The significance of these (except anti-Jo-1) is uncertain.84
latter being associated with a high mortality. A change in voice is a not uncommon manifestation. Juvenile dermatomyositis has also been associated with ischemic ulcerative colitis and celiac disease.58,59
Electromyographic features in polymyositis/dermatomyositis are said to be pathognomonic and include the triad of fibrillation at rest with increased insertional activity and positive sharp waves, polyphasic potentials of short duration and long amplitude and bizarre high-frequency repetitive discharges.57,60
Often stressed in polymyositis/dermatomyositis is the association with an increased risk of developing malignancy.85 Although there has been a great range in reported incidences from small studies, varying from 15% to 60% of cases, recent investigations have suggested that the risk is less.60,86–91 The risk of malignancy appears to be higher among patients with dermatomyositis (23%) than polymyositis (8.9%).92 Polymyositis/dermatomyositis has been described in association with the following malignancies: breast cancer, neuroendocrine carcinoma of the lung, small cell lung cancer, hepatocellular carcinoma, neuroendocrine carcinoma of the liver, duodenal carcinoid, gallbladder carcinoma, carcinoma of the bladder, prostate cancer, renal cell carcinoma, clear cell ovarian carcinoma, fallopian tube cancer, carcinosarcoma of the uterus, nasopharyngeal carcinoma, esophageal cancer, Klatskin tumor (hilar cholangiocarcinoma), thyroid cancer, thymic carcinoma, cancer of the colon, primary gastric melanoma, metastatic melanoma, diffuse large B-cell lymphoma, primary cutaneous B-cell lymphoma-leg type, follicular lymphoma, lymphoplasmocytoid lymphoma, acute myeloid leukemia, and Kaposi sarcoma.93–127 Among the reported associations, breast, stomach, and ovarian tumors are most often cited. In a recent study analyzing patients with dermatomyositis in China, nasopharyngeal cancer was the most frequent association, followed by lung cancer.128 Patients should have a very thorough physical examination combined with routine laboratory investigations, chest X-ray, CT scan of the abdomen and pelvis, and (in female patients) mammography. Underlying malignancy should be suspected in patients who do not respond to therapy or those who develop frequent episodes of myositis.87,129 A recent study suggests that patients requiring more extensive search for malignancy should include those with constitutional symptoms, with rapid onset of dermatomyositis or polymyositis, without Raynaud phenomenon, with a high ESR, and with a very high creatine kinase level.91
The serum usually contains raised levels of creatine kinase, aldolase, lactate dehydrogenase, and transaminases; as not all these may be elevated in any one patient it is usually recommended that all are estimated routinely.6 Sequential muscle enzyme studies are particularly useful for monitoring progress and response to treatment.
Involvement of cardiac muscle is not uncommon and patients may have tachycardia, sinus bradycardia, electrocardiographic abnormalities (e.g., bundle branch block), congestive heart failure, and cardiomegaly.61,62 Restrictive cardiomyopathy has also been described in a patient with dermatomyositis.63 A large population-based study from British Columbia involving 774 patients with inflammatory myopathies revealed a significantly increased risk of myocardial infarction in this group of patients, especially in the first year after diagnosis.64
Pulmonary involvement, as determined by the radiological changes of interstitial fibrosis and/or clinical evidence of impaired respiratory function, may occur in as many as 40% of patients with polymyositis/ dermatomyositis.6 Patients are also at increased risk for development of pulmonary hypertension.65 Pneumomediastinum and interstitial pneumonia are rare complications.66,67 Pulmonary hemosiderosis is an exceptional finding in juvenile dermatomyositis.68 An important recently described association is that between the anti-Jo-1 antibody, pulmonary fibrosis, and dermatomyositis.69–74 More than 50% of patients with anti-Jo-1 antibody have interstitial lung disease.70 Patients with this variant are not at risk of an increased incidence of internal malignancy. Additional features of this variant may include Raynaud phenomenon, arthritis, and tenosynovitis. Spread to the thoracic muscles can result in severe respiratory difficulties; terminal bronchopneumonia is therefore an important cause of death.75
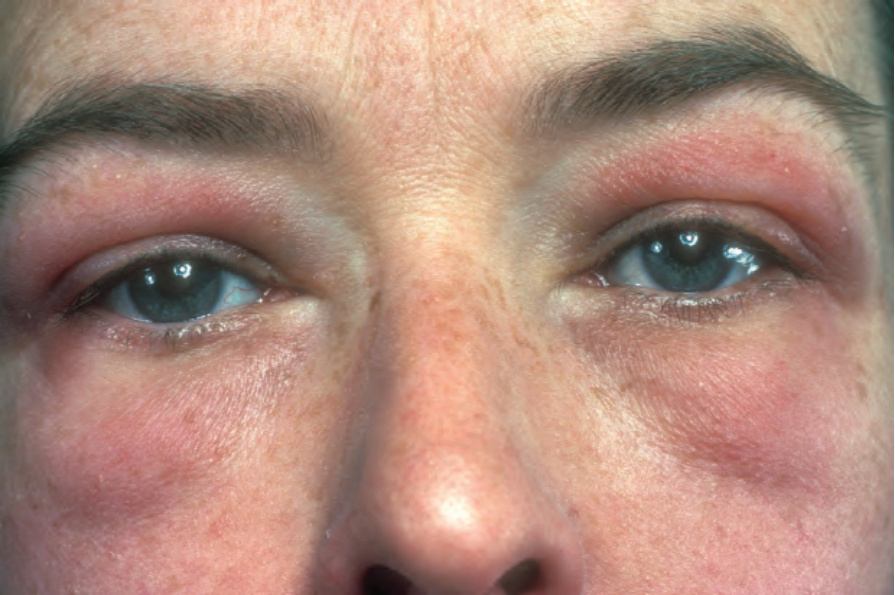
Fig. 17.125 Dermatomyositis: note the characteristic red-mauve discoloration around the eyes. There is also spread onto the cheeks. From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
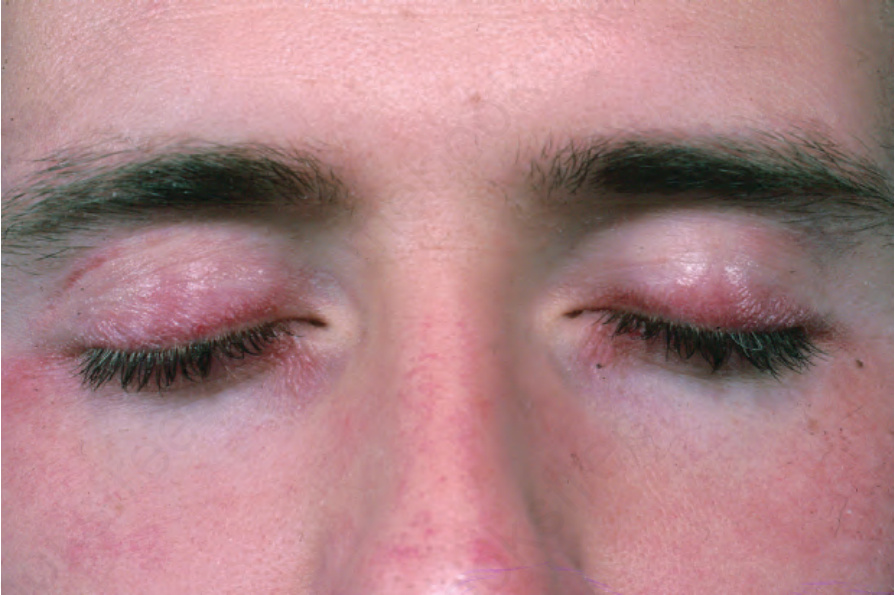
Fig. 17.126 Dermatomyositis: the upper eyelids are particularly affected. From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
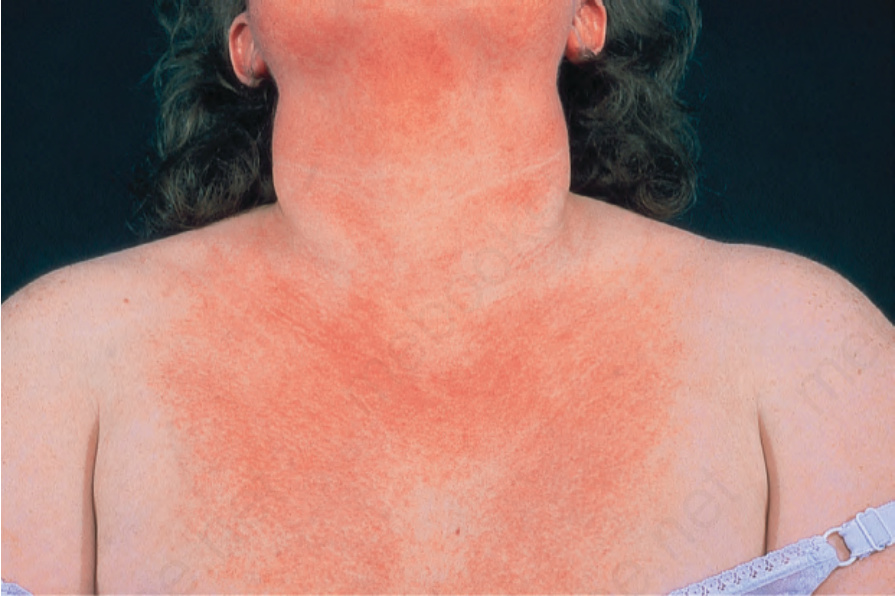
Fig. 17.127 Dermatomyositis: note the erythema and slight scale on this patient’s chest. By courtesy of the Institute of Dermatology, London, UK.
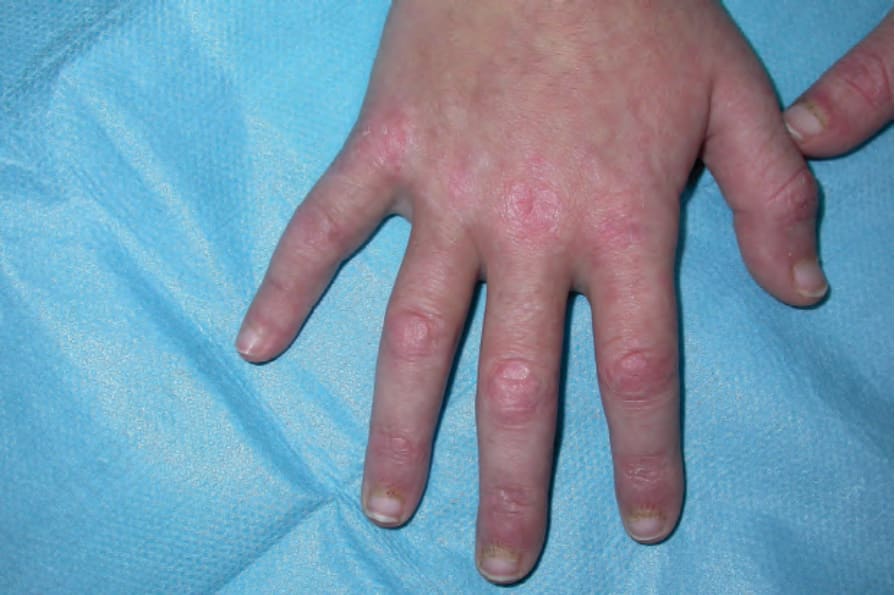
Fig. 17.128 Dermatomyositis: characteristic purple papules on the knuckles (Gottron sign). By courtesy of Dr J.C. Pascual, Alicante, Spain.
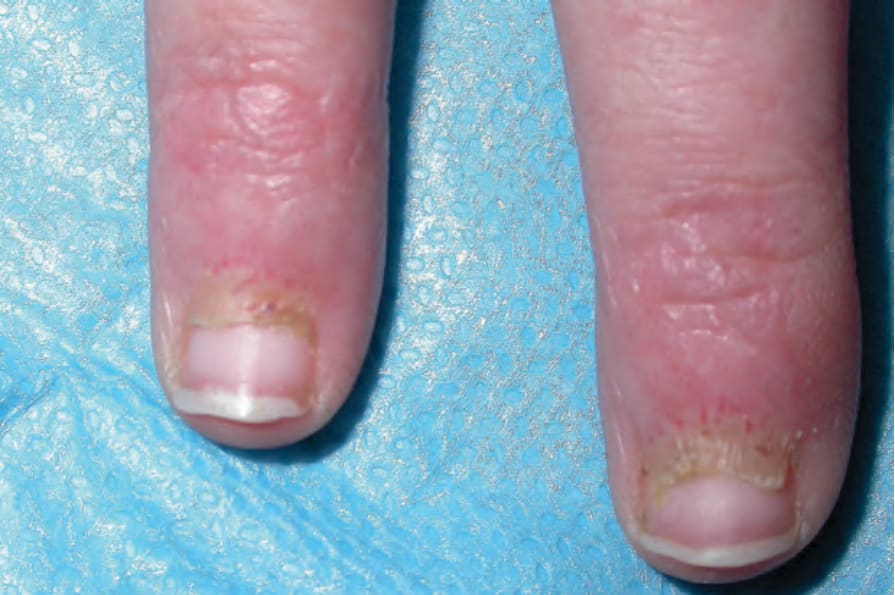
Fig. 17.129 Dermatomyositis: Gottron papules, periungual erythema, and telangiectatic capillary loops. By courtesy of Dr J.C. Pascual, Alicante, Spain.
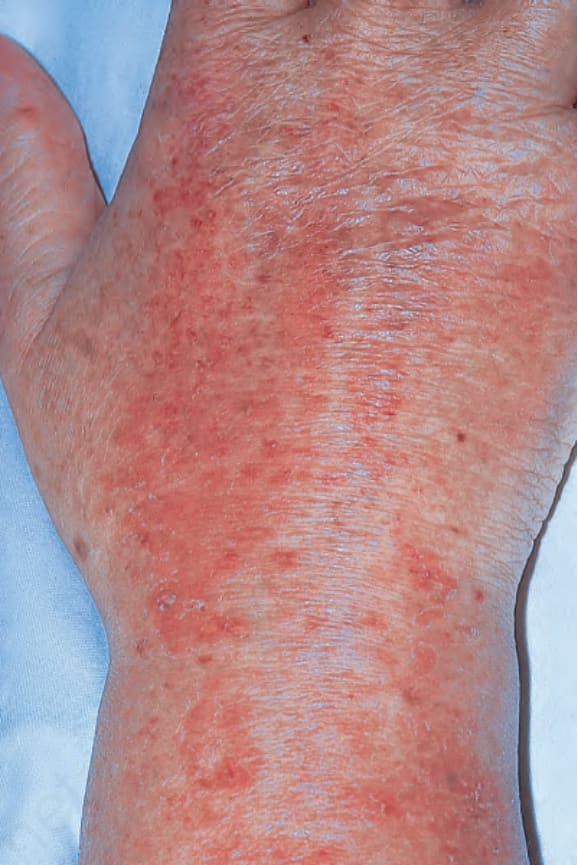
Fig. 17.130 Dermatomyositis: in this patient with longstanding disease, atrophy and variable pigmentary changes (poikiloderma) are present on the dorsum of the hand. By courtesy of the Institute of Dermatology, London, UK.
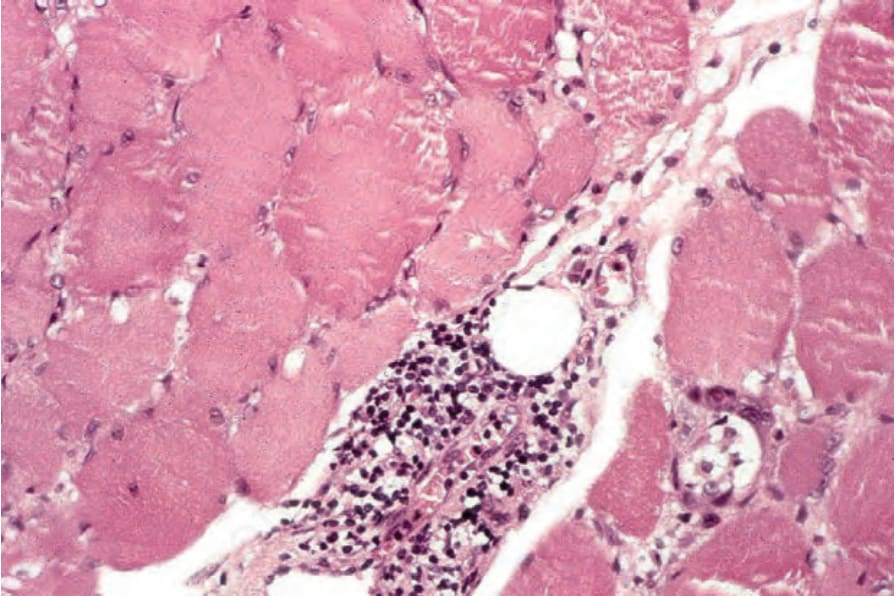
Fig. 17.136 Dermatomyositis: the infiltrate consists predominantly of lymphocytes.

Table 17.12 Diagnostic criteria for polymyositis/dermatomyositis
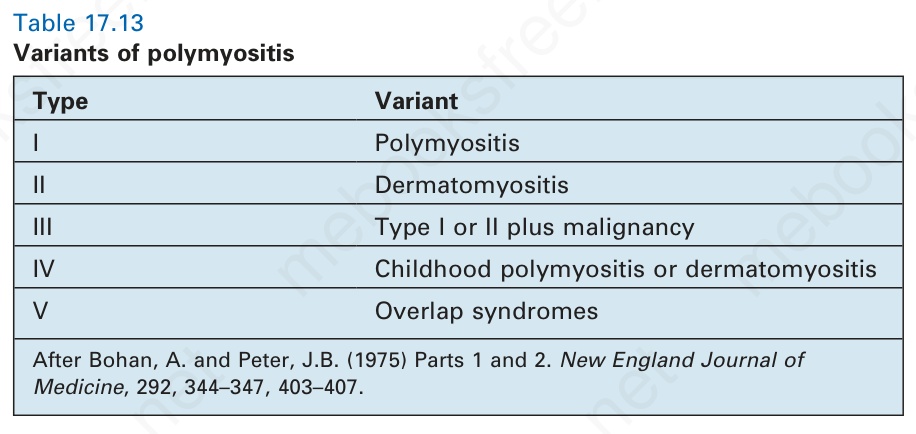
Table 17.13 Variants of polymyositis
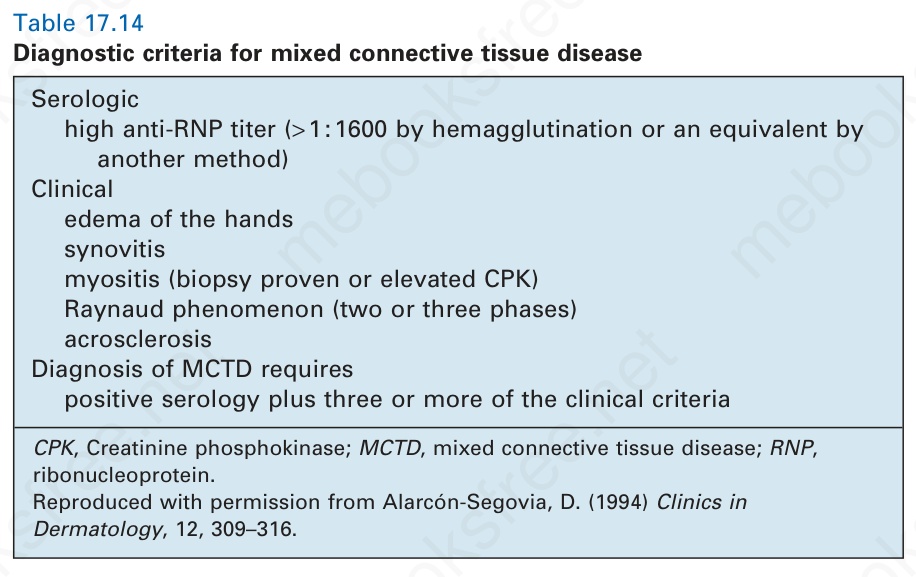
Table 17.14 Diagnostic criteria for mixed connective tissue disease
There appears to be an increased risk of thyroid disease, particularly hypothyroidism, especially in patients with interstitial lung disease.60 Patients with dermatomyositis also have an increased risk for the development of ulcerative colitis.130
Juvenile dermatomyositis, which has an annual incidence of about one new case per million of the population per year, shows a female predominance (2 : 1) and presents most often in the first decade.77 In addition to the features described above there is a high incidence of vasculopathic manifestations, including gastrointestinal ulceration with hemorrhage, which may be fatal.1,131 Multiorgan involvement is common. The condition is often preceded by an infection.132 The prognosis is usually good, with up to 70% of children making a full recovery.132 In severely affected patients,
widespread cutaneous involvement may be complicated by extensive scarring and diffuse calcification.133
Scleroderma/polymyositis overlap (sclerodermatomyositis) is the most common overlap syndrome. Although the myositis component is usually identical to that seen in dermatomyositis/polymyositis, the heliotrope erythema and Gottron papules are usually absent.134 The sclerodermatous cutaneous manifestations tend to be restricted to the peripheries. This overlap syndrome is associated with the Ku antibody, and a case has been reported in association with Graves disease and thrombocytopenic purpura.135 Autoimmune idiopathic thrombocytopenia with anti-Ku antibody has also been associated with dermatomyositis.136
819 Polymyositis/dermatomyositis
Cell-mediated immunity is important in the development of experimental models of polymyositis. Lymphocytes taken from animals with allergic myositis (based upon sequential injections of heterologous muscle with Freund adjuvant) prove cytotoxic to skeletal muscle fibers in culture and may undergo lymphoblastic transformation. Parallels do exist in the human disease, but whether these represent initiating factors or develop as a consequence of muscle damage is unknown.1 A variety of cellular immune abnormalities have been documented, including the presence of activated mononuclear cells within skeletal muscle, abnormal trafficking of mononuclears to skeletal muscle, decreased autologous mixed lymphocyte responses, and mitotic and proliferative responses to autologous muscle.60,152,153
Pathogenesis and histologic features While the etiology and pathogenesis of polymyositis/dermatomyositis are unknown, it has been proposed that environmental factors (e.g., drugs, toxins or viruses) acting in association with a genetic predisposition result in a primarily immune-mediated disorder.60 There is evidence to suggest that both humoral and cell-mediated components are important.
Antinuclear factor is commonly present. Antimyosin and antimyoglobin antibodies have been described, but their significance is uncertain. It is not clear whether they precede or follow the onset of the myositis, and their presence does not explain the cutaneous manifestations. However, antimyosin antibodies accompany any inflammatory myositis and are therefore probably a consequence of muscle necrosis.
There is some evidence to suggest an inherited predisposition with an increased incidence of HLA-B8 and HLA-DR3 in both dermatomyositis and polymyositis, particularly in patients who have anti-Jo-1 antibodies.60,154 There are rare instances of familial disease.60
A number of animal experimental models have shed some light on the possible pathogenesis of human myositis.6 Injection of muscle extracts into a number of animals results in a mild, nonpersistent myositis.60
Several viruses – including Coxsackie B virus, simian acquired immunodeficiency retrovirus, and murine encephalomyocarditis virus – have been shown to induce a chronic myositis-like disease. Virus strain and host genetic factors appear to be of particular importance.60 Although uncertain, it has been suggested that some cases of dermatomyositis, particularly the juvenile variant, may represent an abnormal immunological response to a viral infection.75 Picornaviruses, including the coxsackievirus group, have been particularly implicated.1 The anti-Jo-1 antibody (an antiaminoacyl-tRNA synthetase) reacts with histidyl-transfer RNA synthetase.151 This enzyme has been shown to be capable of interacting with the RNA of a number of picornaviruses in addition to its normal substrate tRNA.60 It has been suggested that the development of the autoantibody may occur as a consequence of this aberrant interaction.60
A further set of antibodies directed against nuclear antigens have been described in 35% to 40% of patients with dermatomyositis/polymyositis:137
• PM-1 (PM-Scl) antibody correlates closely with polymyositis and polymyositis/scleroderma overlap.
• Ku antibody is a marker for sclerodermatomyositis.
• PA-1 antibody correlates with polymyositis, arthritis, and fibrosing alveolitis.
• Mi-2 correlates with dermatomyositis.6,60,137
The presence of antibodies to the RNP antigens, U1 and U2, although not specific, is certainly highly suggestive of dermatomyositis/systemic sclerosis overlap syndrome.135,138,139 Antisignal recognition particle (SRP) antibodies are uncommon, but are usually associated with severe disease.135 Although these antinuclear autoantibodies are of diagnostic value, they have not yet been shown to be of pathogenetic significance.
It is interesting to note that an illness similar to dermatomyositis may be induced by a number of infectious organisms including leishmania, parvovirus (erythrovirus) B19, human immunodeficiency virus, and toxoplasma.155–159 Tuberculous myofasciitis has developed in a dermatomyositis patient.160 Dermatomyositis/polymyositis has been reported as an adverse reaction to a number of drugs, such as hydroxyurea, cyclophosphamide, etoposide, fluvastatin, simvastatin, pravastatin, atorvastatin, ipilimumab, capecitabine, anti-TNF alpha treatment, omeprazole, minocycline, carbimazole, terbinafine, and interferon beta-1a.161–185 Furthermore, polymyositis/dermatomyositis has also been found in association with hepatitis B vaccination, psoriasis, pemphigus foliaceus, Duchenne muscular dystrophy carrier status, familial polyposis colli, ulcerative colitis, hemophagocytic syndrome, organic solvent, and silicone gel-filled breast implants.186–195
Several myositis-specific antibodies have been identified recently, which are closely associated with the disease phenotype. Anti-transcriptional intermediary factor 1 (anti-TIF1) antibodies (previously designated anti-p155-kD protein antibodies) are particularly specific for dermatomyositis in adults and children and have been detected in roughly 30% of both patient groups.140 Furthermore, anti-TIF1 antibodies are strongly associated with malignancies, especially in dermatomyositis patients above the age of 40 years.92 Up to 75% of adult dermatomyositis patients were reported to have developed malignancy.141 Anti-melanoma differentiation antigen 5 (anti-MDA5) antibodies (also designated anti-CADM-140 antibodies) are more frequent in patients with amyopathic dermatomyositis and correlate with interstitial lung disease in Japanese patients.142,143 In addition, it has been demonstrated that anti-MDA5 antibody level correlates with disease activity and that relapse of the disease is associated with re-increase in the levels of anti-MDA5.144 These data suggest that anti-MDA5 antibody levels can be used for monitoring disease activity and as a predictive marker of relapse in patients with amyopathic dermatomyositis.144 Antinuclear matrix protein 2 antibodies (originally designated anti-MJ antibodies) are detected in up to 25% of patients with juvenile myositis and are associated with severe disease course characterized by muscule contractures and atrophy.145,146 Furthermore, the presence of anti-NXP2 autoantibodies significantly increases the risk of calcinosis cutis.147,148 Anti-small ubiquitin-like modifier activating enzyme antibodies are present in less than 10% of patients with adult dermatomyositis and are associated with high incidence of severe dysphagia.140,149
Several studies have confirmed an increased incidence of herpes zoster in dermatomyositis/polymyositis patients in comparison with the healthy controls, especially in females aged older than 50 years with one or more comorbidities, including diabetes, renal disease, obesity, cancer, other autoimmune diseases, MCTDs, vasculitis, and/or treatment with immunosuppressive drugs or corticosteroids.196,197
Dermatomyositis and polymyositis may develop in patients with other known autoimmune disorders, including autoimmune thyroid disease and insulin-dependent diabetes mellitus.60,150 The precise role of humoral immunity in dermatomyositis is unclear, but it is thought to be particularly related to the capillary loss and ischemic damage.151
Direct immunofluorescence of lesional skin reveals granular deposits of immunoglobulin (IgG, IgA, and IgM) and complement at the dermal– epidermal junction in about 35% of patients.198,199 Site selection is of importance, positivity being most frequent with nail bed biopsies.198 A more recent study has demonstrated C5b–9 deposition in blood vessel walls and along the dermal–epidermal junction in conjunction with a negative lupus band test.200,201 This finding has high specificity (93.5%) and sensitivity (78.5%). Epidermal keratinocytes may also be positive for C5b–9 and IgG.201 The finding of C5b–9 in the wall of small blood vessels suggests that a complement-mediated microvascular injury may be of some importance in the pathogenesis of dermatomyositis.
The pathogenesis of childhood dermatomyositis has a predominantly ischemic basis (see below).132
The cutaneous findings are variable. The erythematous eruption shows slight hyperkeratosis and epidermal atrophy, with effacement of the ridge pattern (Fig. 17.131).132 Basal cell liquefactive degeneration is typical and
820 Idiopathic connective tissue disorders
cytoid bodies are sometimes present (Fig. 17.132). Basement membrane thickening is occasionally prominent. There is upper dermal edema and melanophages may be evident. Rarely, the edema results in subepidermal vesiculation.202 A light chronic inflammatory cell infiltrate is usually present (Fig. 17.133). It is commonly restricted to the superficial dermis and is not associated with the cutaneous adnexae. The infiltrate consists of activated T lymphocytes and macrophages with occasional dermal Langerhans cells.203 Helper T cells predominate. In some instances the presence of marked hyperkeratosis, follicular plugging, dermal edema, and increased quantities of basement membrane-like material results in considerable histologic overlap with lupus erythematosus, and clinicopathological correlation is essential. A recent study found the most consistent histologic parameters in dermatomyositis to be vacuolar changes of basal keratinocytes, dermal mucin accumulation, and a mild to moderate dermal mononuclear inflammatory cell infiltrate.204 Dermal sclerosis may occasionally be present.205
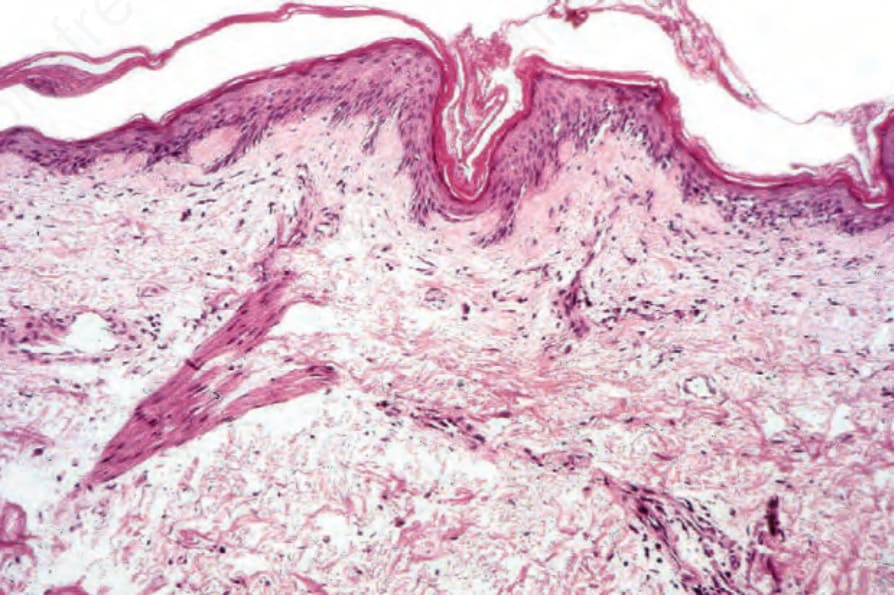
Fig. 17.131 Dermatomyositis: there is hyperkeratosis and epidermal atrophy. Note the mild telangiectasia.
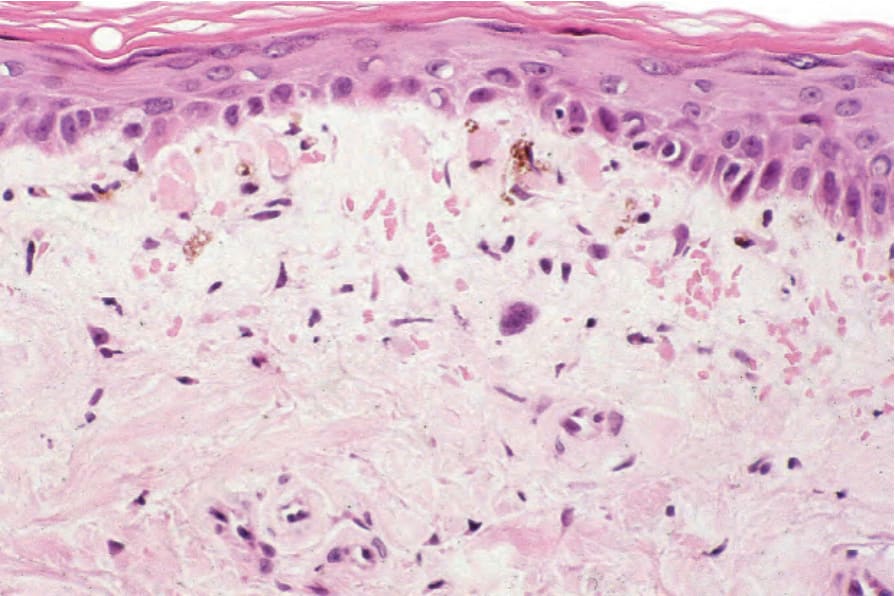
Fig. 17.132 Dermatomyositis: there is atrophy with effacement of the ridge pattern. In this example cytoid bodies are conspicuous. Note the pigmentary incontinence.
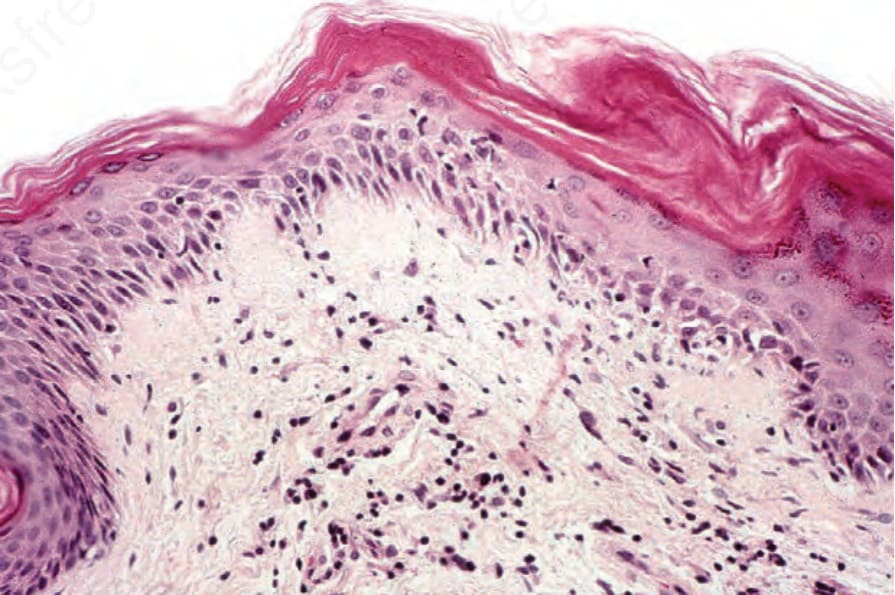
Fig. 17.133 Dermatomyositis: focal, mild basal cell hydropic degeneration is seen on the right. A chronic inflammatory cell infiltrate is present.
incontinence, cytoid body formation, and a patchy lymphocytic inflammatory cell infiltrate. The dermis is edematous, often contains increased mucin, and characteristically shows conspicuous dilated vascular channels. Nuclear atypia of the infiltrate as seen in poikilodermatous mycosis fungoides is not a feature.
Increased quantities of Alcian blue-positive glycosaminoglycans are frequently present within the dermis.202 Sometimes there are foci of calcification and panniculitis is occasionally evident.
The poikilodermatous lesions show hyperkeratosis, mild epidermal atrophy with loss of the epidermal ridge pattern, and basal cell liquefactive degeneration.132 Additional features may include marked pigmentary
Gottron papules are characterized by hyperkeratosis, mild papillomatosis, acanthosis or, less often, epidermal atrophy and the features of interface dermatitis as described above (Fig. 17.134).206,207 The histology of the centripetal flagellate erythema shows the changes of interface dermatitis.51
Ultrastructural studies contribute little to our understanding of dermatomyositis. Tubuloreticular inclusions as described in SLE have been documented in endothelial cell and pericyte cytoplasm, but their significance is uncertain.208
In the juvenile variant, the cutaneous features are similar to those described above with the proviso that fibrosis is sometimes evident, calcification is more common, and occlusive vascular disease (as characterized by fibrous intimal proliferation with fibrin thrombi) is often present.132,133
Skeletal muscle changes include both degenerative and regenerative features in addition to a focal chronic inflammatory cell infiltrate (Figs 17.135 and 17.136).132 The latter is composed predominantly of lymphocytes, but
821 Polymyositis/dermatomyositis
histiocytes, eosinophils, and plasma cells may also be evident.209 The lymphocytes consist of substantial numbers of B cells, particularly in association with blood vessels, in addition to T cells, which are predominantly found in and around the altered muscle fibers.210 As in cutaneous lesions, T-helper cells predominate. Up to 25% of muscle biopsies may, however, show no evidence of inflammation.4 The degenerative fibers are swollen and intensely eosinophilic and may show loss of striations (Fig. 17.137). Some fibers are vacuolated, but others appear granular or fragmented (Fig. 17.138). Proliferation and centralization of muscle nuclei is common, as is sarcoplasmic basophilia – features of regeneration (Fig. 17.139). Histologic changes identical to inclusion body myositis, namely rimmed vacuoles, have also recently been reported in patients with dermatomyositis.211,212
If material from a longstanding ‘burned out’ lesion is biopsied, the muscle fibers are atrophic and there is endomysial fibrosis. Perifascicular atrophy – the presence of one or two rows of atrophic fibers at the edge of a fascicle – is said to be characteristic of dermatomyositis.213 The muscle pathology in dermatomyositis and polymyositis is said to differ.60 In dermatomyositis the inflammatory cell infiltrate tends to be septal or perivascular, whereas in polymyositis it is intrafascicular. Muscle necrosis in dermatomyositis tends to involve small groups of fibers, while in polymyositis the affected fibers tend to be single and sparse.
Denervation neuropathic features are also occasionally present, presumably due to involvement by the inflammatory process of small intramuscular
822 Idiopathic connective tissue disorders
nerve fibers.132 Steroid atrophy of type II muscle fibers may be seen in biopsies from treated patients.
In childhood dermatomyositis, vascular changes affecting the capillaries, venules, and arterioles are common.214 The inflammatory component, which is usually quite sparse, consists of lymphocytes, monocytes, and plasma cells centered predominantly on the vasculature in the perifascicular connective tissue.215 Muscle changes are variable and range from perifascicular atrophy in milder disease through to focal necroses and infarction in the more seriously affected patients, in whom fibrosis may also be a feature.215 The vascular lesions include endothelial cell swelling and necrosis with or without occlusion, non-necrotizing lymphocytic vasculitis, and loss of the peripheral fascicular capillary bed.215
Serologic
high anti-RNP titer (> 1 : 1600 by hemagglutination or an equivalent by
another method) Clinical
edema of the hands synovitis myositis (biopsy proven or elevated CPK) Raynaud phenomenon (two or three phases) acrosclerosis Diagnosis of MCTD requires
Immunofluorescence of muscle biopsies in childhood dermatomyositis commonly shows vascular intramural IgM and C3.215
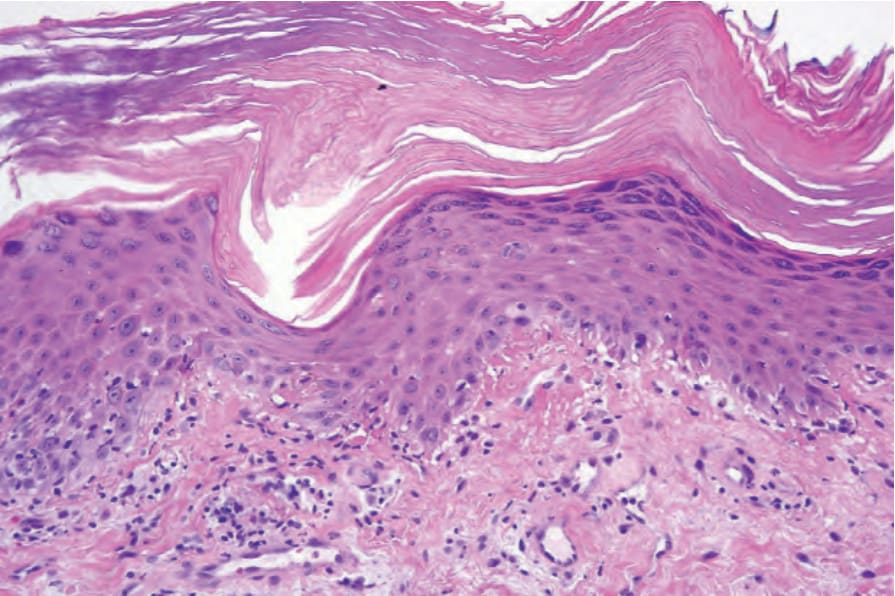
Fig. 17.134 Gottron papule: note the hyperkeratosis, hypergranulosis, and irregular acanthosis simulating lichen planus. There is basal cell hydropic degeneration and cytoid bodies are present in the superficial dermis. By courtesy of D. Whittemore, DO, MD Anderson Cancer Center, Houston, Texas, USA.
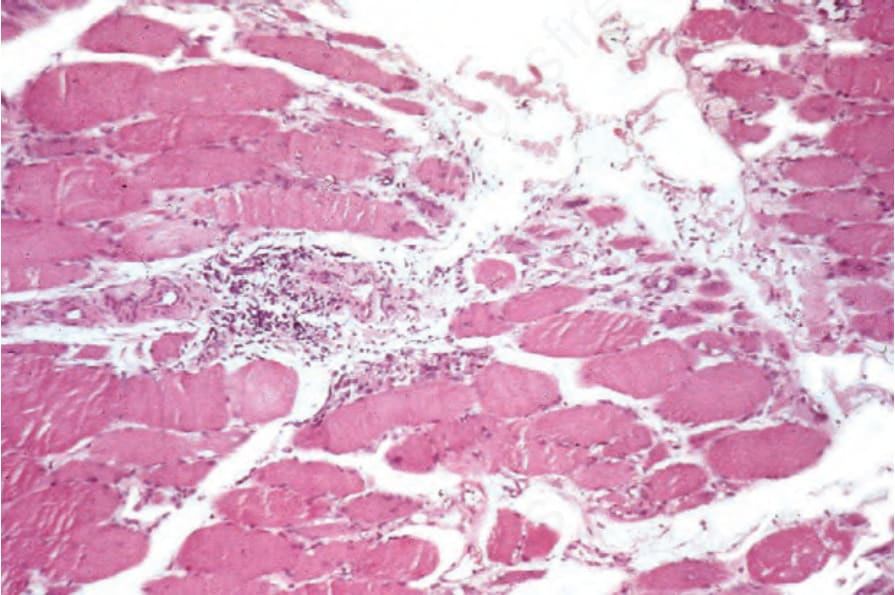
Fig. 17.135 Dermatomyositis: note the perivascular chronic inflammatory cell infiltrate.
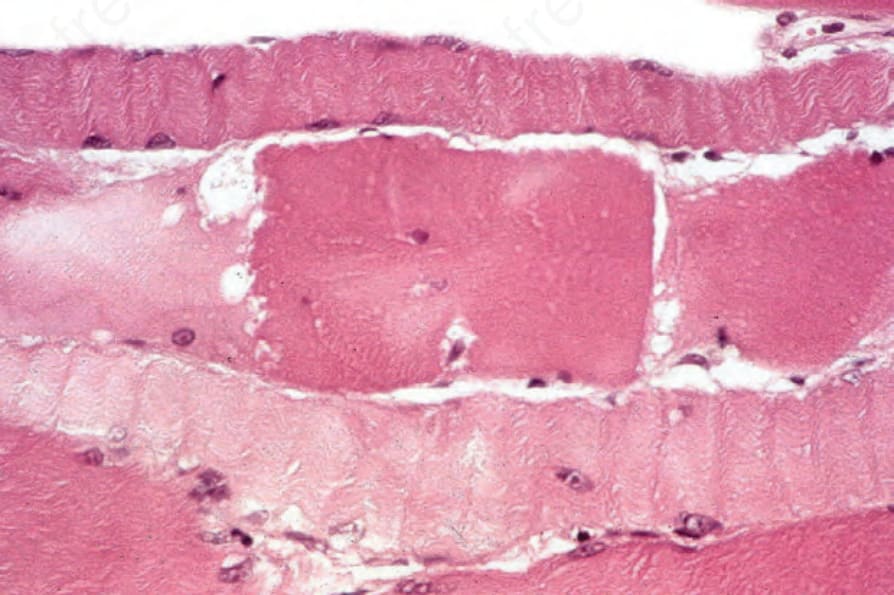
Fig. 17.137 Dermatomyositis: the central fiber is intensely swollen, eosinophilic, and fragmented; there is a loss of striations.
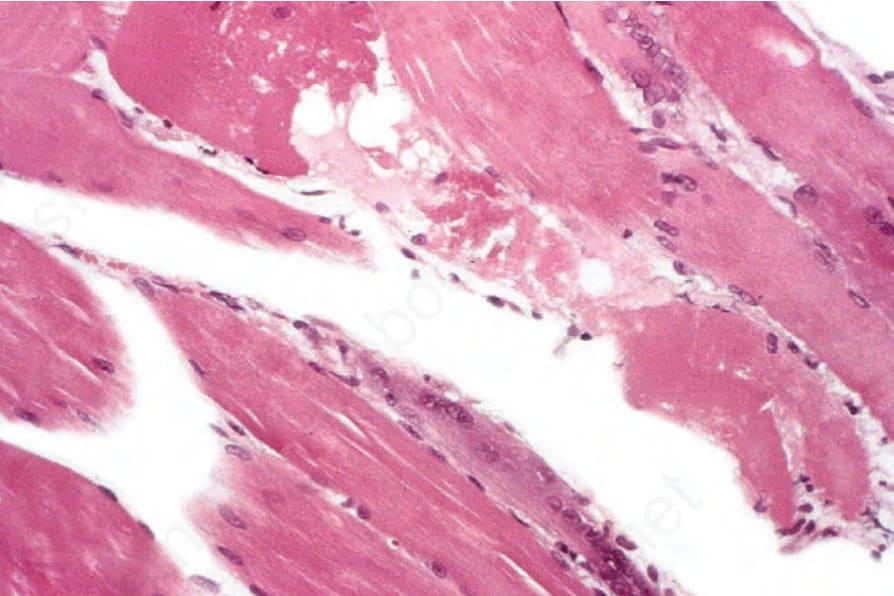
Fig. 17.138 Dermatomyositis: the fiber in the upper midfield is swollen, eosinophilic, vacuolated, and in places granular; beneath is a regenerating basophilic cell.
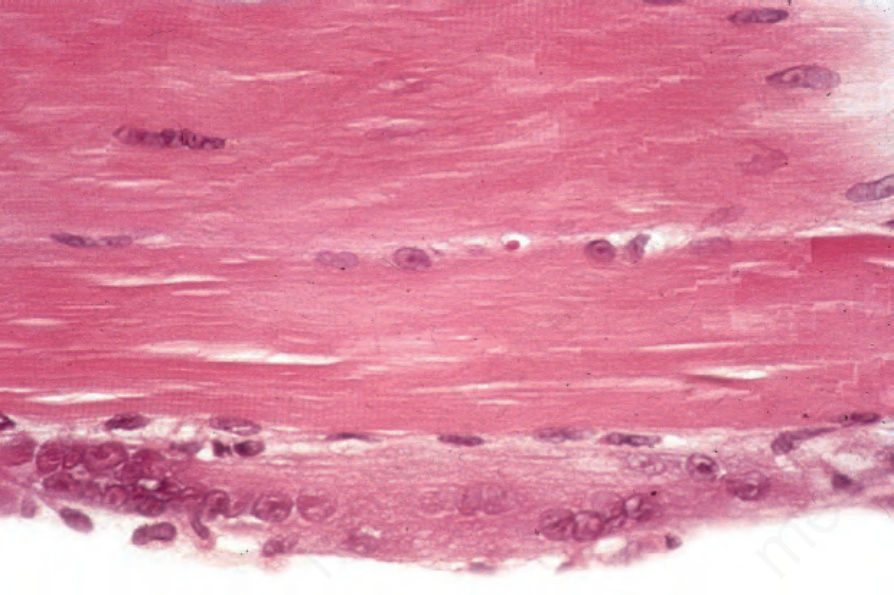
Fig. 17.139 Dermatomyositis: the lower fiber is basophilic and shows excessive nuclei – features of regeneration. Note the centralization of nuclei in the upper fiber.