疾病定義與分類
- 典型結節性多動脈炎 (classic polyarteritis nodosa;Kussmaul-Maier disease) 是一種罕見的全身性血管炎,主要侵犯中型與小型動脈。有些人認為此病並非一個獨立疾病 (disease sui generis),而是較廣義地視為一種具有多種誘發原因與疾病關聯的症候群。
- 典型 polyarteritis nodosa 在臨床與組織學上均與顯微鏡下多血管炎 (microscopic polyangiitis;microscopic polyarteritis nodosa、microscopic polyarteritis) 有重疊,但 polyarteritis nodosa 主要為中型血管之血管炎,而 microscopic polyangiitis 主要為小血管血管炎,後者於他處討論。
臨床特徵 (Clinical Features)
典型結節性多動脈炎 (Classical polyarteritis nodosa)
-
典型 polyarteritis nodosa 是一種多系統疾病,臨床表現變化多端 (protean)(表 16.5)。應注意 1990 年美國風濕病學會 (American College of Rheumatology) 的標準並未區分 polyarteritis nodosa 與 microscopic polyarteritis。然而,在較新的 Chapel Hill 共識命名中,二者依所侵犯之血管大小區分(見表 16.1)。
-
polyarteritis nodosa 即使在使用皮質類固醇 (corticosteroids) 治療下,仍伴有顯著的罹病率與死亡率。經治療後,存活率約在 75% 至 80% 之間。
-
雖然可能影響廣泛年齡層,但患者最常見於第五或第六個十年 (decade)。男性較好發 (4:1)。患者常以全身性症狀 (constitutional symptoms) 表現,包括體重減輕、發熱 (pyrexia) 與食慾不振。
-
皮膚病灶常見,出現於 30% 至 60% 的患者。最常見的是可觸及的紫斑性病灶 (palpable purpuric lesions) 與潰瘍灶,尤其侵犯下肢(圖 16.46–16.48)。網狀青斑 (livedo reticularis) 亦為常見的皮膚表現(圖 16.49)。亦可見皮膚結節。斑丘疹 (maculopapular rash)、水疱形成與膿疱性病灶為偶見特徵(圖 16.50–16.53)。
-
關節侵犯(關節痛與關節炎)常見;關節炎通常為不對稱性,尤其侵犯下肢。非特異性肌肉疼痛與無力為附加特徵。常見肌肉萎縮 (muscle wasting)。
-
周邊與中樞神經系統 (CNS) 侵犯均常見。前者表現為感覺神經病變(麻木或感覺異常 paresthesias)、運動神經病變(垂腕 wrist drop 或垂足 foot drop),以及合併之感覺運動病灶(多發性單神經炎 mononeuritis multiplex 與多發性神經病變 polyneuropathy)。CNS 侵犯可表現為意識混亂、定向力障礙或譫妄 (delirium)。眼部侵犯為 polyarteritis nodosa 的罕見特徵。併發症包括脈絡膜梗塞 (choroidal infarction)、缺血性視神經病變 (ischemic optic neuropathy)、視網膜動脈阻塞 (retinal artery occlusion)、表層鞏膜炎 (episcleritis)、潰瘍性角膜炎 (ulcerative keratitis)、葡萄膜炎 (uveitis) 與眼窩假性腫瘤 (orbital pseudotumor)。
-
腎臟侵犯常見,且具重大意義,因其後遺症 — 腎衰竭與高血壓 — 是本病最常見的死亡原因之一。患者偶有因腎梗塞 (renal infarction) 引起的腰痛發作。典型 polyarteritis nodosa 患者常出現高血壓,部分患者可能進入惡性高血壓期 (malignant phase)。因此,檢測蛋白尿、血尿與紅血球柱 (red cell casts) 的尿液分析,以及血清肌酸酐 (serum creatinine) 估計,為必要的早期檢查。
-
胃腸道侵犯亦為重要的罹病與死亡原因。症狀包括噁心、嘔吐與腹痛。嚴重併發症包括胃腸道出血、穿孔與梗塞,後者為不罕見的死亡原因。亦可見肝膽道 (hepatobiliary tract) 侵犯。膽囊與胰臟侵犯亦曾被報告,可為偶然發現,或患者可能以急性膽囊炎 (acute cholecystitis) 症狀表現。
-
心臟侵犯發生於不到三分之一的病例。表現包括心包炎 (pericarditis)、心律不整,以及因冠狀動脈侵犯所致之心肌梗塞(圖 16.54)。雖然常稱 polyarteritis nodosa 不侵犯肺部,但在例外病例中可見肺部侵犯,患者偶爾抱怨氣喘、咳血 (hemoptysis) 與積液 (effusions)。雖然肺部臨床侵犯罕見,但屍檢評估顯示侵犯支氣管動脈 (bronchial arteries) 的動脈炎並不罕見,在某一小型系列中佔 70%。
-
睪丸炎 (orchitis),通常為單側,是 polyarteritis nodosa 的特徵性表現。受影響患者以急性睪丸炎症狀,或提示睪丸腫瘤的特徵表現。
-
實驗室檢查常顯示貧血、白血球增多 (leukocytosis) 與 ESR 上升。有時可見低效價 (low-titer) 的類風濕因子 (rheumatoid factor) 與抗核抗體 (antinuclear antibody),偶有患者可檢出冷球蛋白 (cryoglobulin)。亦可檢出血清補體 (serum complement) 濃度降低。ANCAs 在典型 polyarteritis nodosa 患者中不常見。一個具豐富經驗的研究團隊估計,典型本病患者中具有 ANCAs 者不到 5%。然而,ANCAs 的存在應強烈考慮為 ANCA 相關性血管炎 (ANCA-related vasculitis) 而非 polyarteritis nodosa。兒童 polyarteritis nodosa 可能以兩種形式表現:嬰兒型 (infantile variant),可能與川崎病 (Kawasaki disease) 相關;以及兒童型 (childhood form),與成人 polyarteritis nodosa 相似(亦見川崎病一節)。
皮膚型結節性多動脈炎 (Cutaneous polyarteritis nodosa)
-
除典型 polyarteritis nodosa 外,亦曾描述「局部型(皮膚型)結節性多動脈炎」(‘localized (cutaneous) polyarteritis nodosa’)。這是一種相對良性的變異型,患者發生皮膚病灶(常持續非常長的時間),但依定義從不出現嚴重的內臟侵犯。在一項研究中,79 名皮膚型 polyarteritis nodosa 患者經平均 6.9 年追蹤,無人發展為全身性血管炎。可發生於任何年齡,包括兒童期,且無性別好發傾向。本病偶爾與 minocycline 治療相關。
-
患者有反覆發作,期間發生壓痛、疼痛性結節,尤其在小腿,但有時可能相當廣泛。個別病灶直徑由 2 mm 至 2 cm 不等。早期為粉紅或紅色,較成熟的結節可能呈紫色調。患者有時亦表現網狀青斑 (livedo reticularis),通常在小腿,且常與成群結節相關。其他併發症包括潰瘍,以及罕見的壞疽 (gangrene)。極偶爾地,患者發生類似白色萎縮 (atrophie blanche) 的病灶。
-
其他特徵包括發熱、倦怠、關節痛與肌痛,且周邊神經可能受影響,但從不出現更廣泛內臟侵犯的證據。
-
免疫螢光 (immunofluorescence) 常顯示皮膚動脈壁內有 IgM 及/或補體,提示可能的免疫複合體 (immune complex) 致病機轉。罕見報告指出皮膚型多動脈炎母親所生之嬰兒亦發病,並隨後緩解,提示存在致病性循環因子 (pathogenic circulating factor)。
致病機轉與組織學特徵 (Pathogenesis and histologic features)
-
polyarteritis nodosa 的致病機轉所知甚少。根據血清免疫複合體濃度、免疫螢光研究與超微結構研究,有人提出典型 polyarteritis nodosa 為免疫複合體媒介 (immune-complex mediated)。然而,在許多患者中無法證實免疫複合體,其在本病發展中的角色具有爭議。重要的可疑抗原包括 B 型肝炎病毒 (hepatitis B virus, HBV) 表面抗原與冷球蛋白 (cryoglobulins)。已顯示相當數量的 polyarteritis nodosa 患者有循環 HBV 抗原。此外,在偶見患者中已鑑定出含 HBV 抗原與免疫球蛋白的循環免疫複合體。在少數患者的受侵犯血管內亦已鑑定出 HBV 表面抗原。法國曾報告 HBV 相關的 polyarteritis nodosa 病例減少,並有人提出此現象是疫苗接種計畫的結果。然而,罕見情況下,polyarteritis nodosa 亦可能在 B 型肝炎疫苗接種後發生。人類免疫缺乏病毒 (human immunodeficiency virus) 感染亦曾於 polyarteritis nodosa 或類 polyarteritis nodosa 症候群病例中被報告。
-
部分患者已記錄有 C 型肝炎病毒 (hepatitis C virus) 感染證據。在一項研究中,20% 的患者具有針對 C 型肝炎病毒的抗體。微小病毒 (parvovirus) 感染在偶見病例中與 polyarteritis nodosa 相關。
-
在兒童期 polyarteritis nodosa 中,似乎與 A 群鏈球菌 (group A streptococci) 有顯著的關聯。
-
雖然有些證據提示感染期間產生的免疫複合體扮演某種角色,但在許多病例中無法證實此關係。因此,典型 polyarteritis nodosa 的致病機轉在許多患者中仍不明。
-
在一小部分患者中,本病與腺苷脫胺酶 2 缺乏 (deficiency of adenosine deaminase 2, DADA2) 相關。這是一種近期描述的自體發炎性疾病,繼發於第 22q11.1 號染色體上 CECR1 的突變。其特徵為早期兒童期發病的類網狀青斑性血管病變 (livedoid vasculopathy),伴有 CNS 侵犯與輕度免疫缺乏。患者常具有與 polyarteritis nodosa 無法區分的臨床與組織病理學表現,反映出多重致病途徑的概念。其早期發病與傳統 polyarteritis nodosa 形成鮮明對比。因此,在嬰兒與幼童遇到的病例中,應考慮調查此種可能的遺傳疾病。
-
典型與局部型 polyarteritis nodosa 兩種變異型之皮膚病灶的組織學特徵相似,且變化多端。在某些情況下,其變化與侵犯淺層真皮血管的白血球碎裂性血管炎 (leukocytoclastic vasculitis) 無法區分(圖 16.55)。然而更具特徵的是侵犯深層真皮或皮下脂肪之肌性動脈 (muscular arteries) 的壞死性血管炎 (necrotizing vasculitis);這些變化亦見於內臟,常伴有梗塞(圖 16.56)。雖然血管壁的整個圓周與厚度常受影響,但有時變化為局灶性。polyarteritis nodosa 的典型特徵是血管變化呈不連續性,受影響節段之間有未受侵犯的跳躍性病灶 (skip lesions)(圖 16.57)。
-
急性變化,即纖維素樣壞死 (fibrinoid necrosis),侵犯肌層 (muscle coat),常破壞內彈性膜 (internal elastic lamina);此點常以彈性組織染色最能呈現(圖 16.58)。壞死伴隨著由嗜中性球、嗜酸性球與單核細胞組成的發炎細胞浸潤。白血球碎裂 (leukocytoclasis) 有時為附加特徵。血栓形成常見,並可能因表面上皮的缺血性壞死而複雜化。癒合中的病灶伴隨纖維母細胞增生 (fibroblastic proliferation) 與最終的纖維性瘢痕化。在癒合期,常出現淋巴球性浸潤而非嗜中性球性浸潤。內膜動脈炎 (endarteritis) 常明顯可見,且內彈性膜的任何破壞均為永久性。在含多條血管的楔形切片中,常見的特徵性表現為存在處於不同演化階段的病灶。深部、外科切開式切片 (surgical incisional biopsies) 對於診斷 polyarteritis nodosa 的皮膚侵犯是必要的。打孔切片 (punch biopsy) 常無法取樣到典型受侵犯的較大血管。此外,因本病的多灶性 (multifocal) 性質,診斷易受取樣誤差影響。有時可在顯微鏡下察見動脈瘤 (aneurysm) 形成。
-
內臟侵犯基於壞死性動脈炎 (necrotizing arteritis) 的效應。有趣的是,結節狀腫脹(動脈瘤 aneurysms)更為明顯。其效應取決於梗塞與出血的相對交互作用。典型 polyarteritis nodosa 的腎臟侵犯主要由大血管血管炎所致,伴隨血栓與梗塞,加上高血壓的效應(圖 16.59)。患者亦可能表現局灶性、節段性增生性或壞死性腎絲球腎炎 (focal, segmental proliferative or necrotizing glomerulonephritis),類似於 microscopic polyarteritis nodosa 患者所見(圖 16.60)。
鑑別診斷 (Differential diagnosis)
- 典型 polyarteritis nodosa 與 microscopic polyangiitis 之區別,基於受侵犯血管的大小、器官侵犯的範圍與類型,以及 ANCAs 的存在。與 polyarteritis nodosa 相似,硬結性紅斑 (erythema induratum) 常顯示中型血管的血管炎,但相對於 polyarteritis nodosa,其另有伴隨的小葉性脂膜炎 (lobular panniculitis)。皮膚淋巴球性血栓性/斑狀淋巴球性動脈炎 (cutaneous lymphocytic thrombophilic/macular lymphocytic arteritis) 可能代表 polyarteritis nodosa 的癒合期,而非一個獨立的疾病實體。
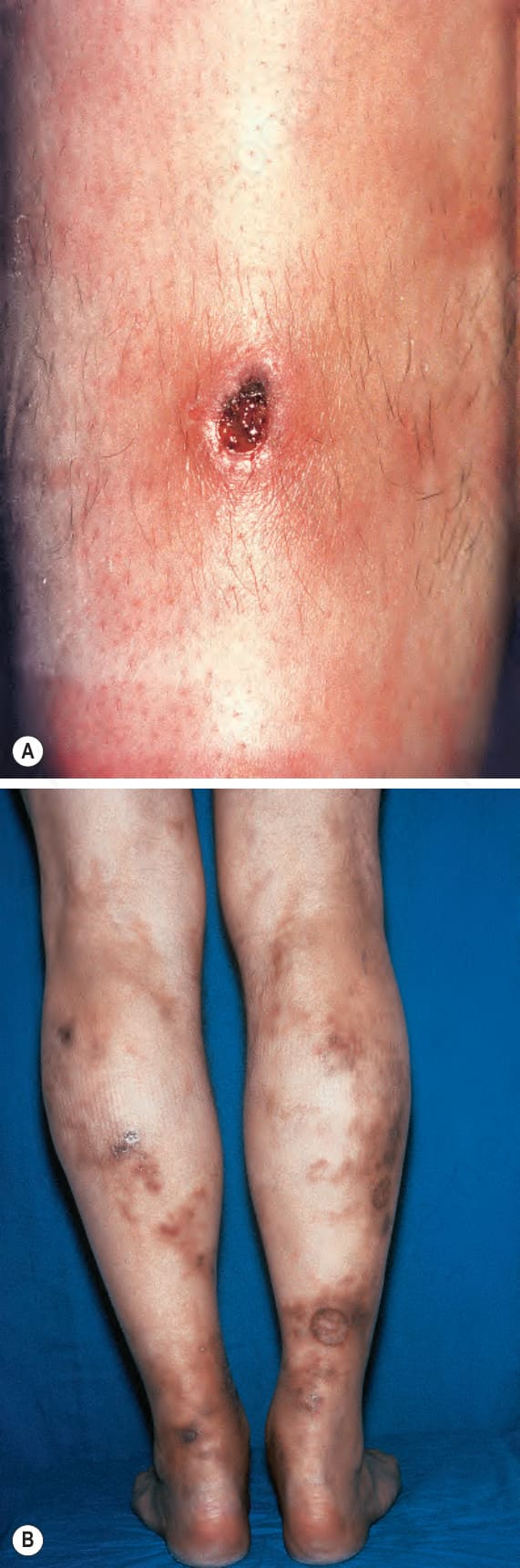
圖 16-46:結節性多動脈炎:(A) 脛部界線分明、邊緣硬結呈紫色的潰瘍;(B) 多發性潰瘍、結節與網狀青斑 (livedo reticularis) 灶。 (Polyarteritis nodosa) By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
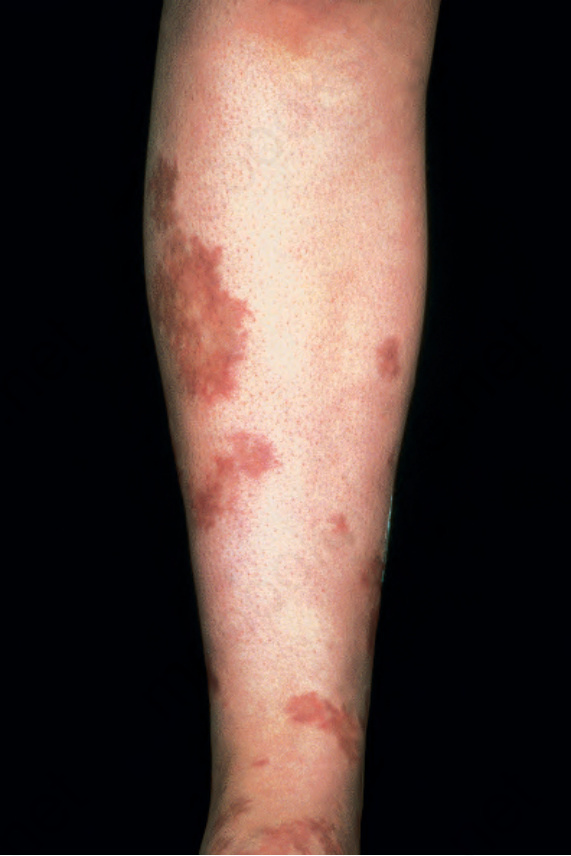
圖 16-47:結節性多動脈炎:此患者以雙腿大片出血性病灶 (hemorrhagic lesions) 表現。 (Polyarteritis nodosa) By courtesy of the Institute of Dermatology, London, UK.
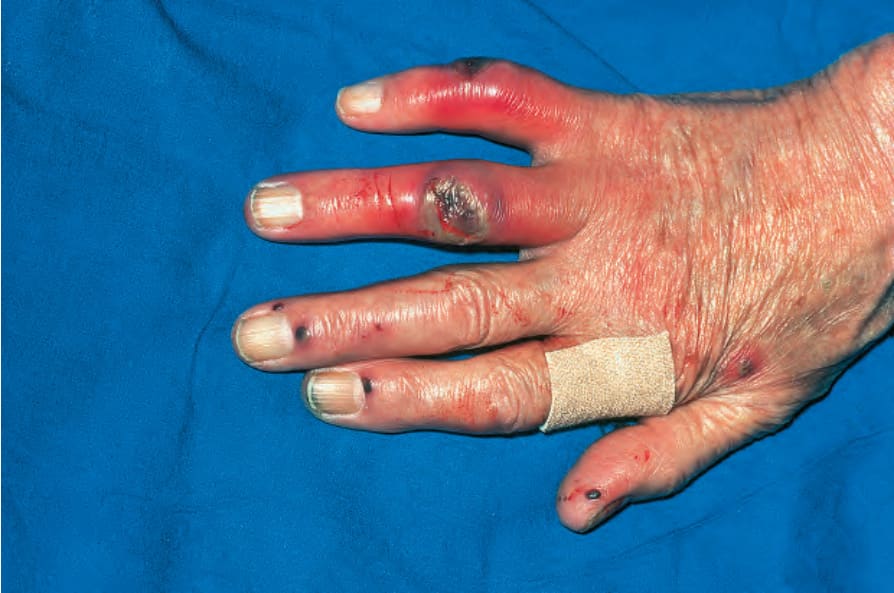
圖 16-48:結節性多動脈炎:表皮梗塞 (epidermal infarction) 導致這些指(趾)端潰瘍 (digital ulcers)。 (Polyarteritis nodosa) By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
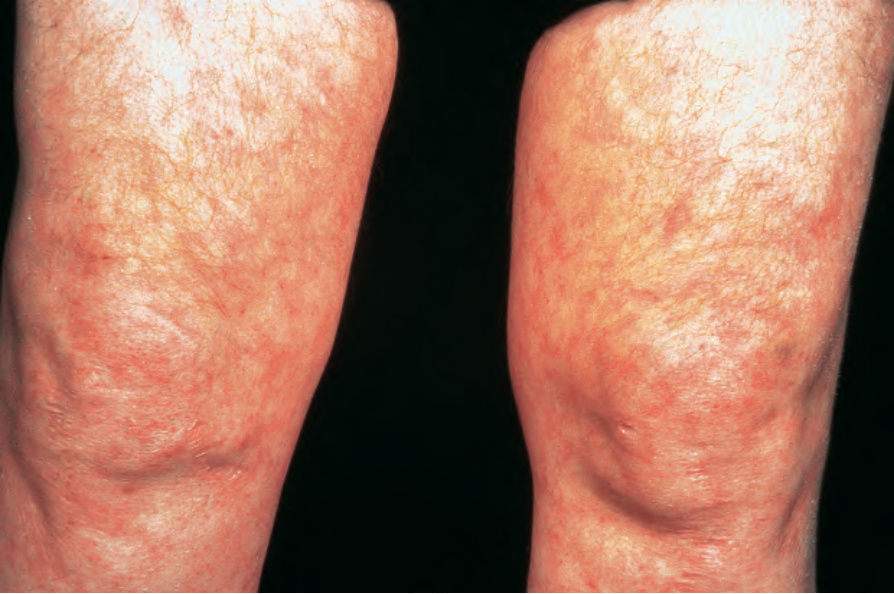
圖 16-49:結節性多動脈炎:此患者顯示明顯的網狀青斑 (florid livedo reticularis)。 (Polyarteritis nodosa) By courtesy of the Institute of Dermatology, London, UK.
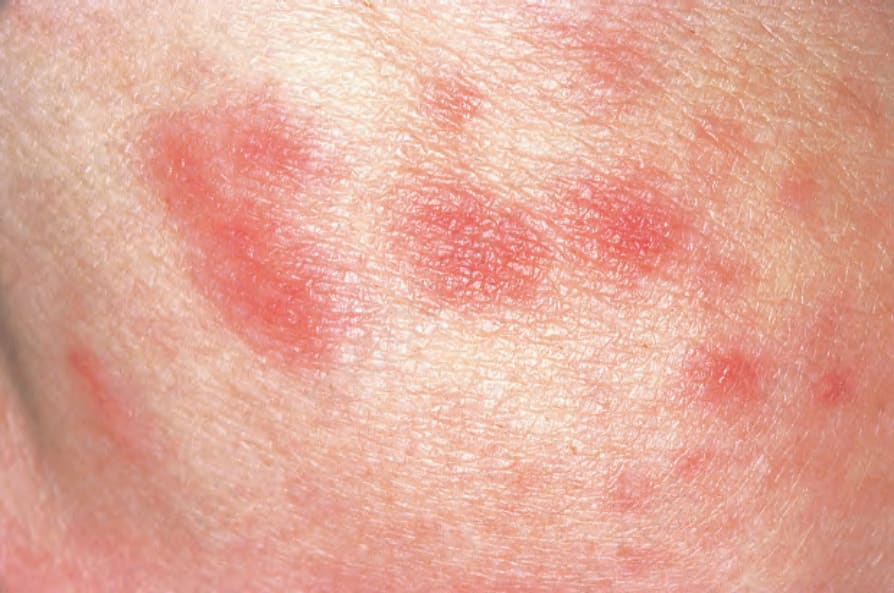
圖 16-50:結節性多動脈炎:偶爾可見紅斑性斑疹 (erythematous macules)。 (Polyarteritis nodosa) By courtesy of the Institute of Dermatology, London, UK.
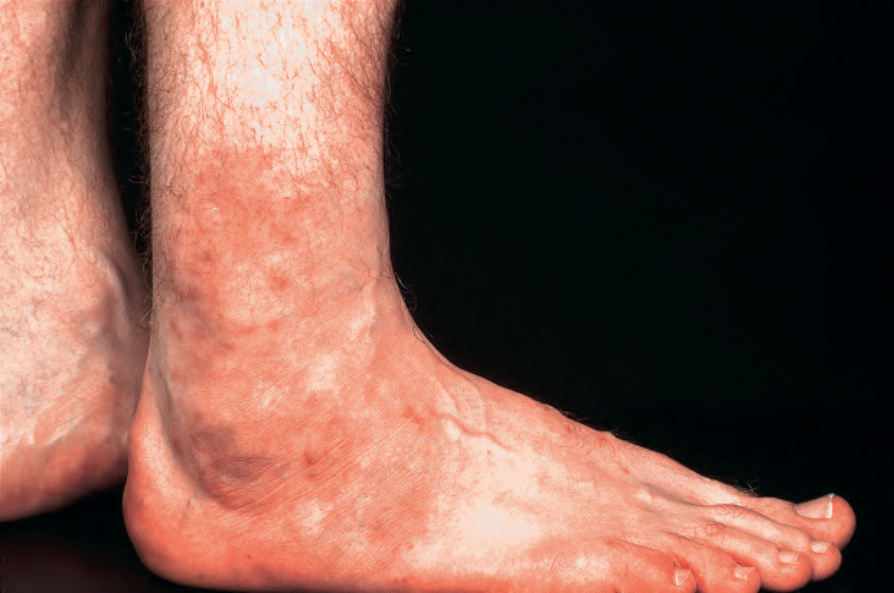
圖 16-51:結節性多動脈炎:此患者踝部周圍出現紅斑性丘疹 (erythematous papules)。 (Polyarteritis nodosa) By courtesy of the Institute of Dermatology, London, UK.
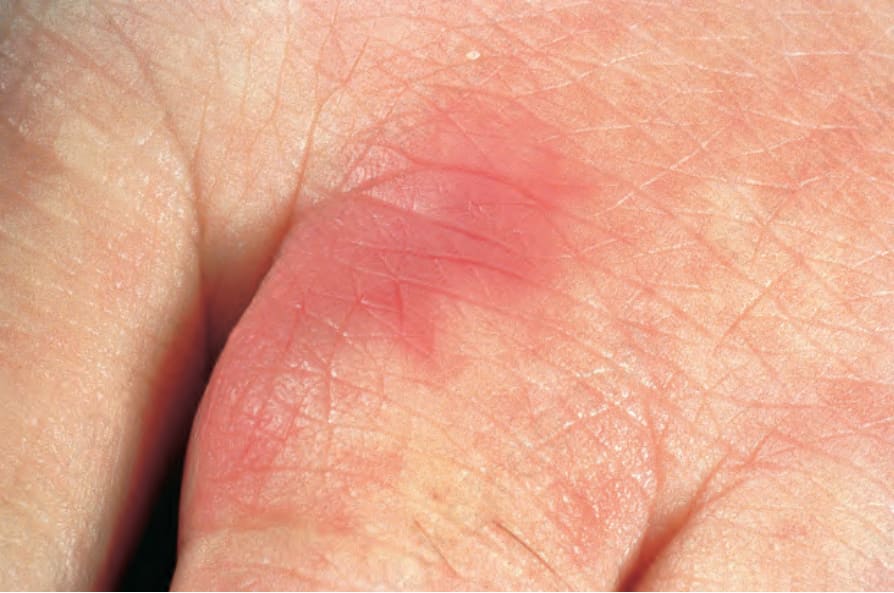
圖 16-52:結節性多動脈炎:此患者以肢端紅斑性病灶 (acral erythematous lesions) 表現。 (Polyarteritis nodosa) By courtesy of the Institute of Dermatology, London, UK.
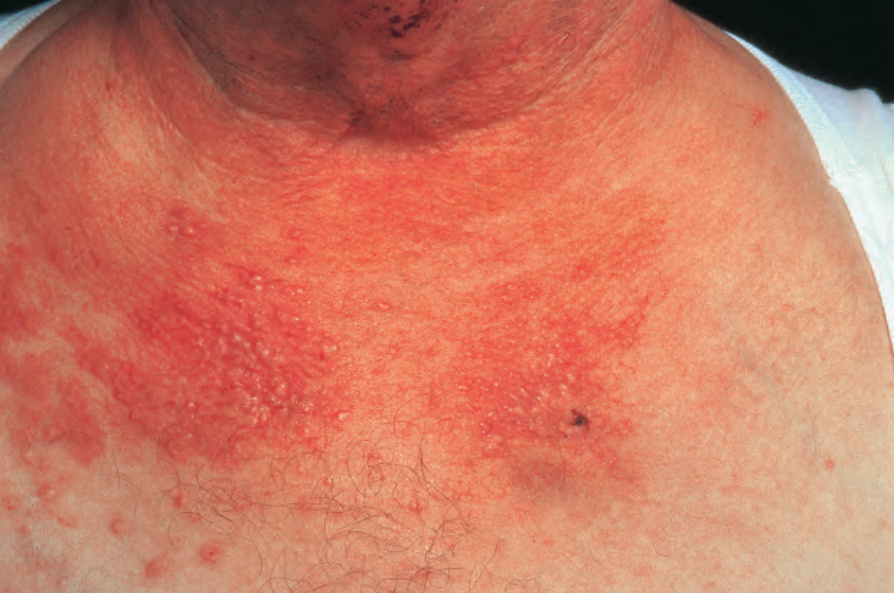
圖 16-53:結節性多動脈炎:在某些患者中,強烈的嗜中性球浸潤 (neutrophil infiltrate) 導致膿疱性病灶 (pustular lesions),如此患者所見。 (Polyarteritis nodosa) By courtesy of the Institute of Dermatology, London, UK.
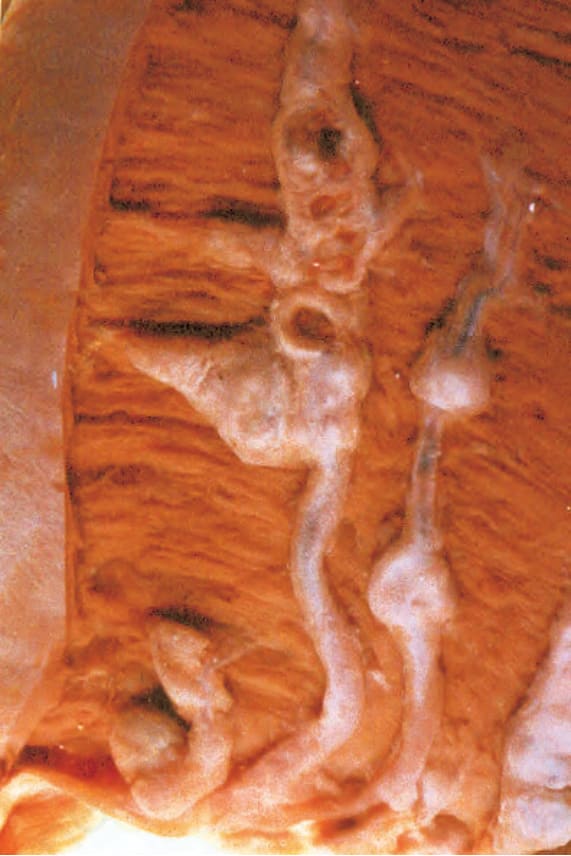
圖 16-54:結節性多動脈炎:顯示明顯動脈瘤樣擴張 (aneurysmal dilatation) 的冠狀動脈,現今已非常罕見(博物館標本)。 (Polyarteritis nodosa) By courtesy of the Department of Pathology, St Thomas’ Hospital, London, UK.
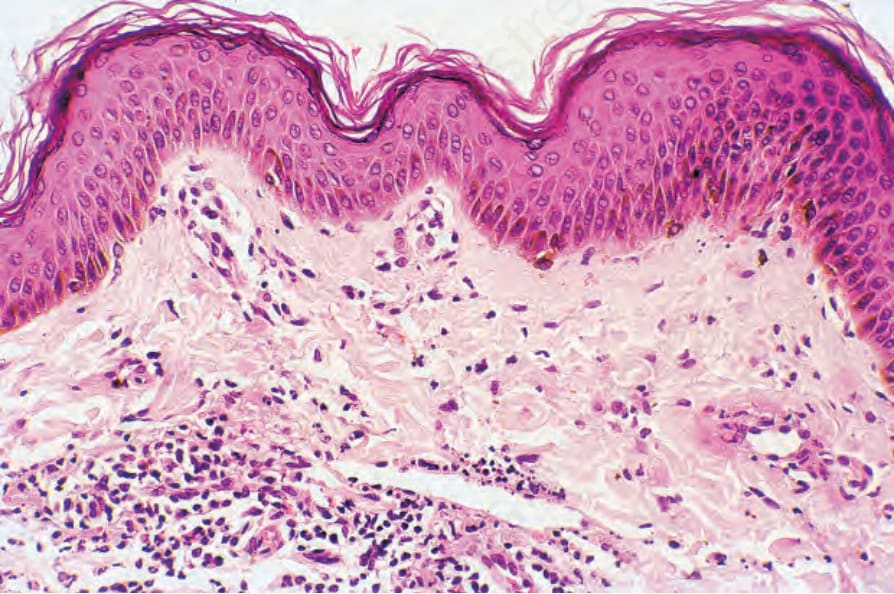
圖 16-55:結節性多動脈炎:本例的特徵為淺層白血球碎裂性血管炎 (superficial leukocytoclastic vasculitis)。重要的是須記得,此組織學病灶可能代表嚴重的全身性疾病。 (Polyarteritis nodosa)
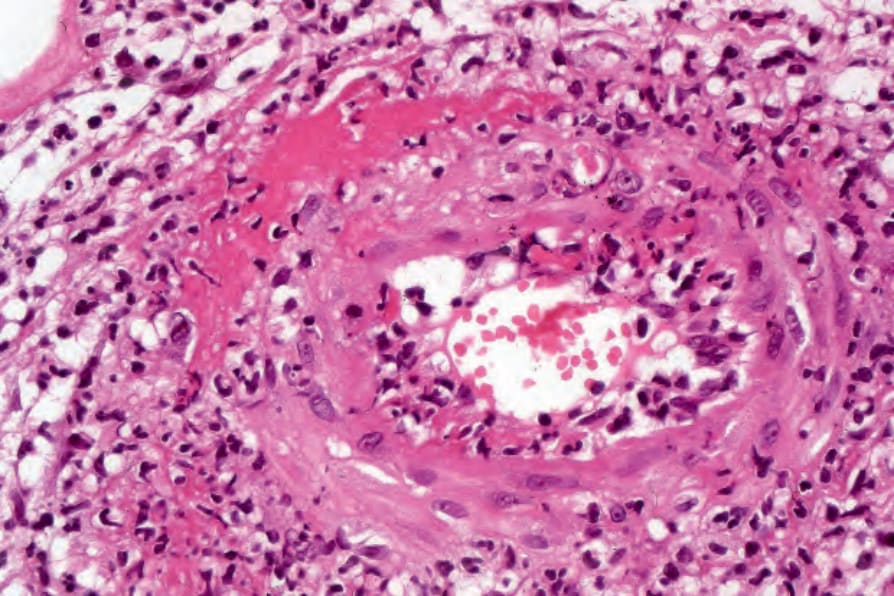
圖 16-56:結節性多動脈炎:高倍視野顯示纖維素樣壞死 (fibrinoid necrosis)。 (Polyarteritis nodosa)
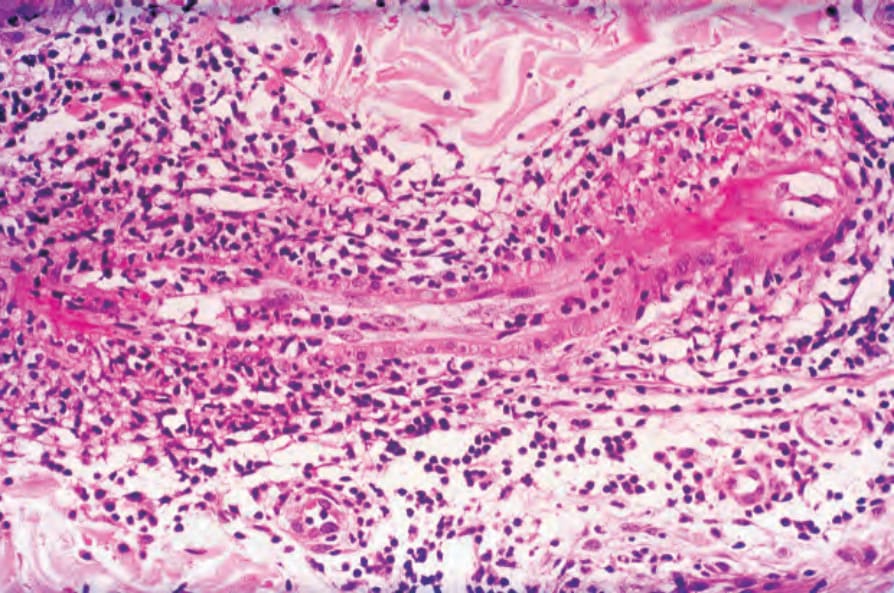
圖 16-57:結節性多動脈炎:纖維素樣壞死 (fibrinoid necrosis) 侵犯此血管節段的兩側末端,而中段相對未受影響。 (Polyarteritis nodosa)
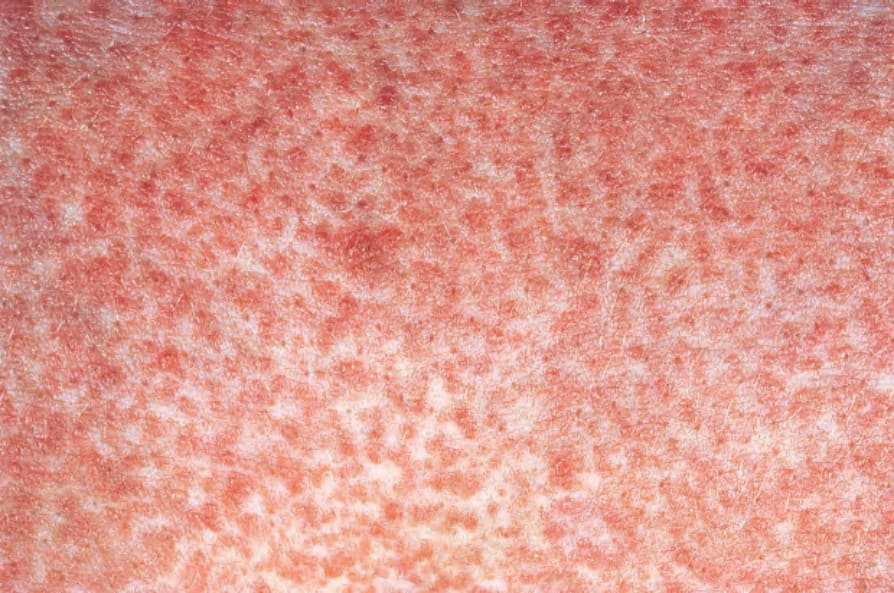
圖 16-61:川崎病 (Kawasaki disease):紅斑性斑狀疹 (erythematous macular eruption)。 By courtesy of W.G. Phillips, MD, Institute of Dermatology, London, UK.
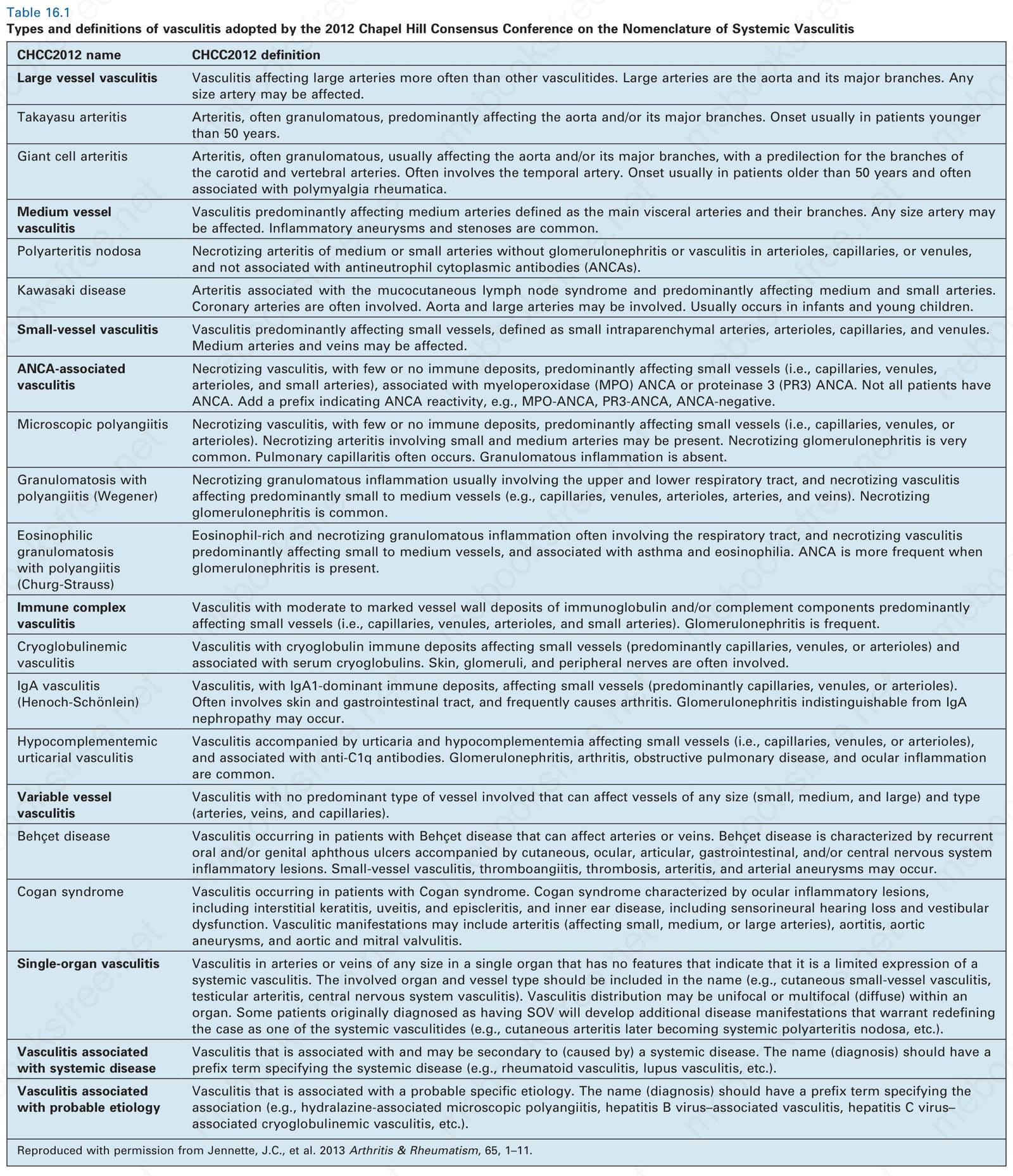
表 16-1:2012 年 Chapel Hill 全身性血管炎命名共識會議 (2012 Chapel Hill Consensus Conference on the Nomenclature of Systemic Vasculitis) 所採用之血管炎類型與定義。
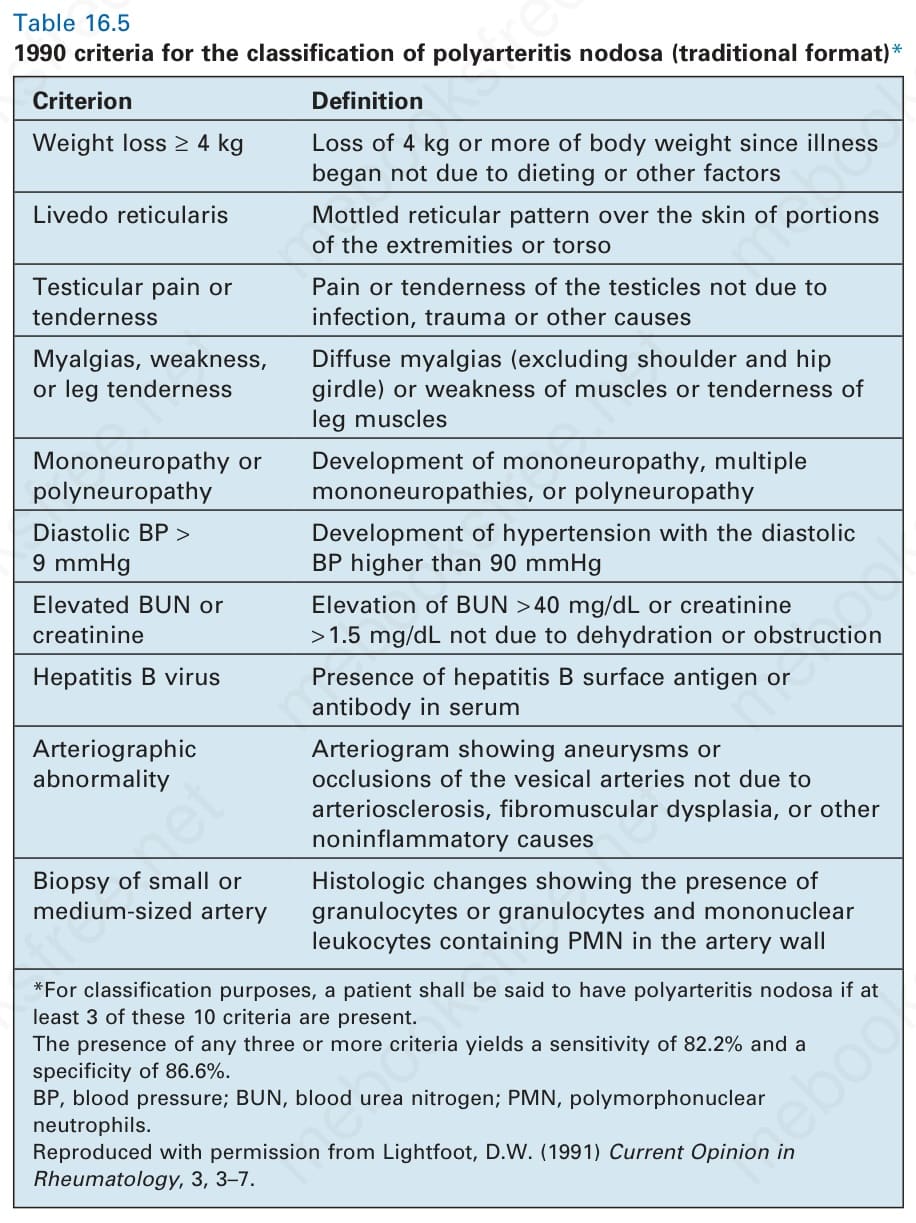
表 16-5:1990 年結節性多動脈炎分類標準(傳統格式)(1990 criteria for the classification of polyarteritis nodosa, traditional format)。

表 16-6:川崎症候群 (Kawasaki syndrome):診斷指引。
川崎病(黏膜皮膚淋巴結症候群)(Kawasaki disease, mucocutaneous lymph node syndrome)
- 發熱持續 ≥ 5 天
- 多形性皮疹 (polymorphous rash)
- 雙側結膜充血 (bilateral conjunctival injection)
- 至少一項下列黏膜變化:– 唇部紅斑或皸裂 – 草莓舌 (strawberry tongue) – 口腔與咽部黏膜瀰漫性充血
- 急性非化膿性頸部淋巴結病變(至少一顆淋巴結 ≥ 1.5 cm)
- 至少一項下列周邊肢端變化:– 手掌與足底紅斑 – 手足硬結性水腫 (indurative edema) – 指尖膜狀脫屑 (membranous desquamation)
發熱加上上述四項標準必須存在方可確立診斷;須排除其他可能以相似臨床表現出現的疾病。Reproduced with permission from Wortman, D.W. (1992) Seminars in Dermatology, 11, 37–47.
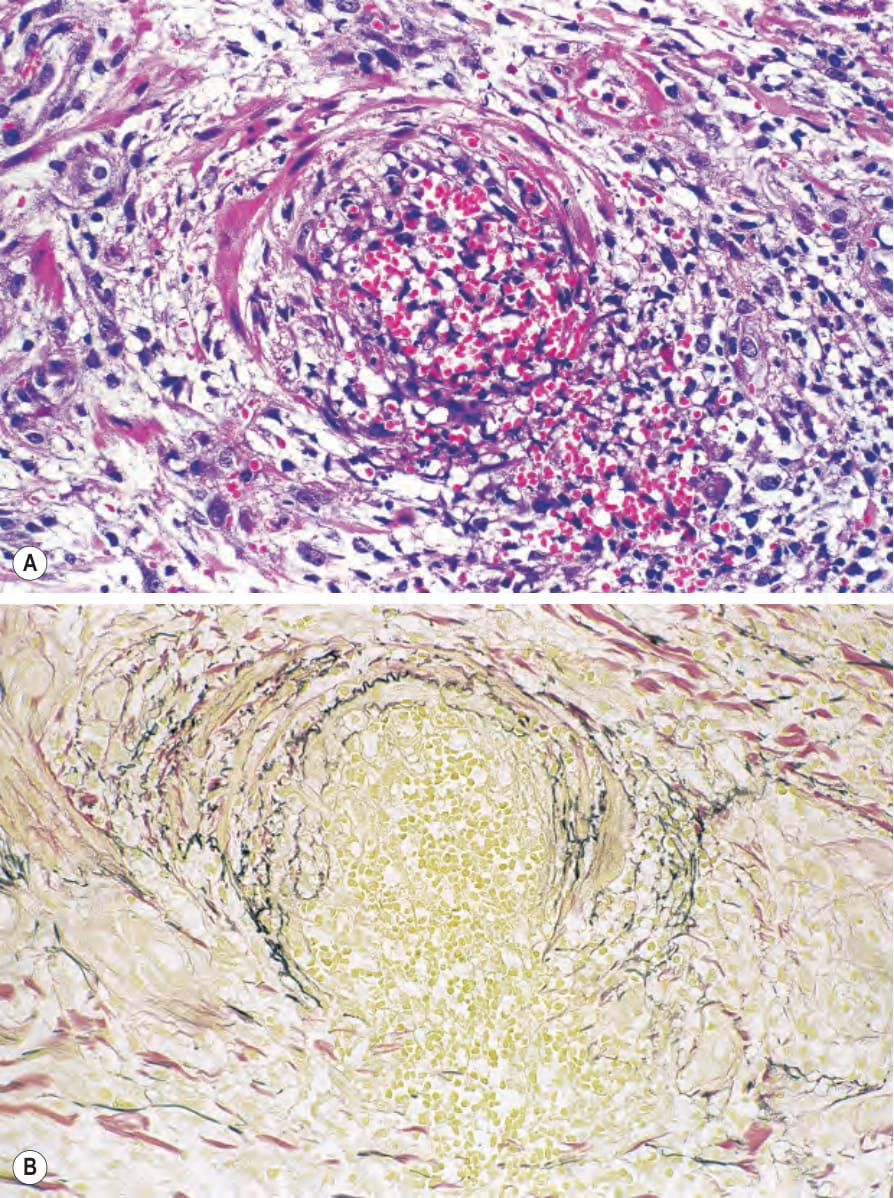
圖 16-58:結節性多動脈炎:(A) 有明顯的紅血球外滲 (red cell extravasation);(B) elastic–van Gieson 染色顯示內彈性膜 (internal elastic lamina) 的破壞。 (Polyarteritis nodosa)
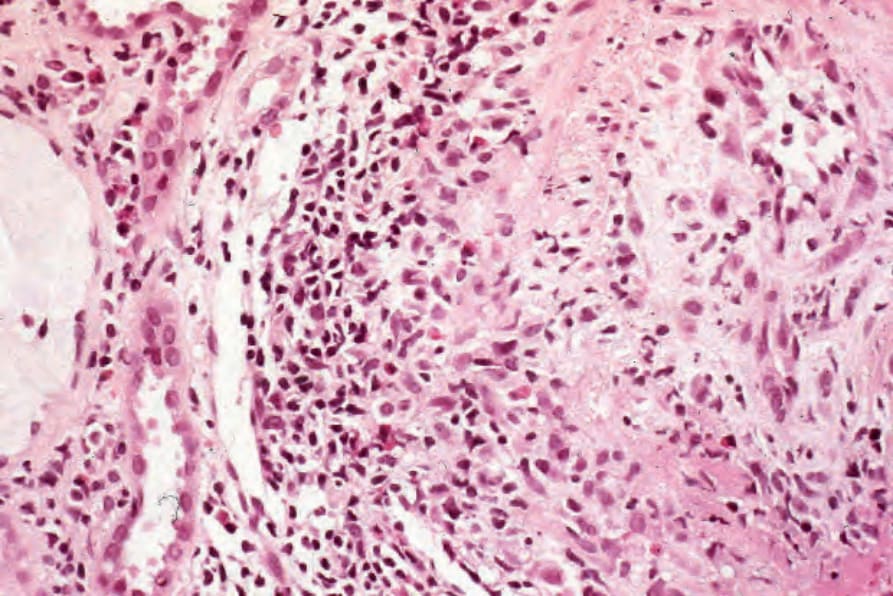
圖 16-59:結節性多動脈炎:在此腎臟切片中,一條弓狀動脈 (arcuate artery) 顯示壞死性血管炎 (necrotizing vasculitis) 與纖維內膜增厚 (fibrointimal thickening)。發炎細胞浸潤含有明顯的嗜酸性球。 (Polyarteritis nodosa)
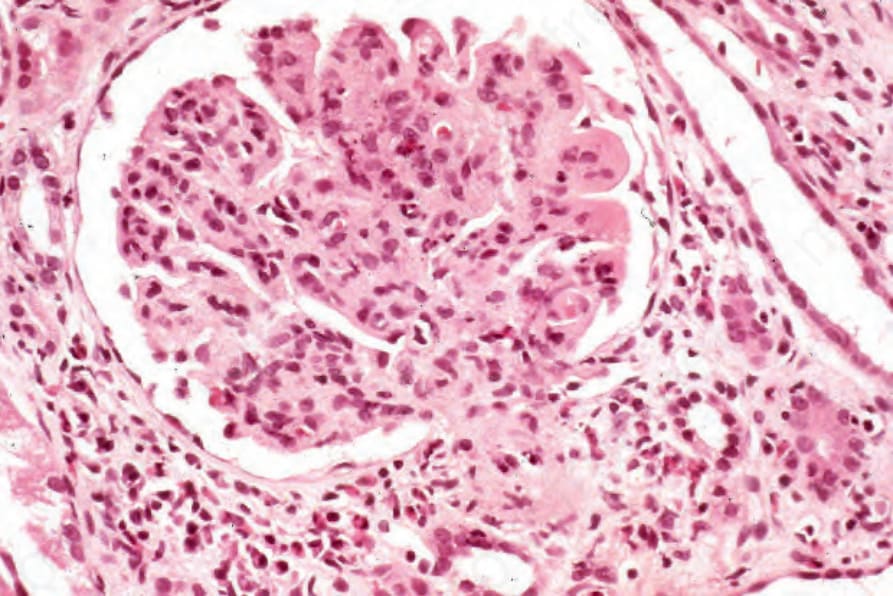
圖 16-60:結節性多動脈炎:節段性壞死性腎絲球腎炎 (segmental necrotizing glomerulonephritis)。 (Polyarteritis nodosa)