12-遺傳性家族性類澱粉沉積症 (Heredofamilial Amyloidoses)
家族性地中海熱 (Familial Mediterranean Fever)
臨床特徵 (Clinical Features)
- 這是一種體染色體隱性遺傳 (autosomal recessive) 的自體發炎性疾病 (autoinflammatory disease)。
- 可分為兩種表現型:第 1 型與第 2 型。第 1 型的特徵為陣發性發燒、漿膜炎 (serositis)、腹膜炎 (peritonitis)、滑膜炎 (synovitis),少數情況下出現心包膜炎 (pericarditis) 與腦膜炎 (meningitis)。反覆的發炎發作可導致類澱粉沉積症 (amyloidosis)。在第 2 型中,病人初始即以 amyloidosis 表現,其餘則無症狀。
- 皮膚病灶罕見,包括 Henoch-Schönlein 紫斑 (Henoch-Schönlein purpura) 與下肢類似丹毒 (erysipelas) 的紅斑。
- 也可能出現脂膜炎 (panniculitis)、反覆性蕁麻疹 (recurrent urticaria)、結節性多動脈炎 (polyarteritis nodosa)、類乾癬 (psoriasis-like) 病灶、水疱性皮膚病灶 (bullous skin lesions)、血管周圍淋巴球性皮膚炎 (perivascular lymphocytic dermatitis) 與類肉瘤病 (sarcoidosis)。
- 也曾記載甲褶 (nail fold) 微血管異常,表現為微血管袢 (capillary loops) 的迂曲增加與擴大。
- 尚未有皮膚 amyloid 沉積的描述。
致病機轉與組織學特徵 (Pathogenesis and Histologic Features)
-
Familial Mediterranean fever 是由位於第 16 號染色體 16p13 上的 MEFV 基因突變所引起,該基因編碼 pyrin,一種參與去活化 (deactivating) 免疫反應的蛋白質。
-
pyrin 的缺陷導致 interleukin-1 過度生成,造成促發炎狀態 (proinflammatory state),進而促成 amyloid 的形成。
-
在 familial Mediterranean fever 中,由一種血清前驅蛋白形成此病的 amyloid。此前驅物是一種 HDL,稱為血清類澱粉蛋白 A (serum amyloid A)。
-
具有良好的敏感度與特異度(約 70–90%),但其他研究在腹部脂肪墊切片 (abdominal fat pad biopsies) 中顯示出較差的敏感度。
-
雖然明顯的臨床病灶通常不是次發性類澱粉沉積症 (secondary amyloidosis) 的特徵,但有時可在正常皮膚的檢體中發現小型沉積。通常這些沉積位於血管周圍,但偶爾也可能出現在真皮的其他部位,甚至在皮下脂肪中。也可見 amyloid 沉積於汗腺周圍。據稱這些沉積呈灶性 (focal),而腹部皮下脂肪 (abdominal subcutaneous fat) 被推薦為最可能呈陽性的部位。血液透析相關類澱粉沉積症 (Hemodialysis-associated amyloidosis) 是次發性 amyloidosis 的一種特殊型態,將於下文描述。
-
類丹毒 (erysipelas-like) 病灶的特徵為血管周圍由淋巴球、組織球 (histiocytes) 與嗜中性球組成的混合性浸潤,並伴有白血球碎裂 (leukocytoclasia)。雖未見血管炎 (vasculitis),但在直接免疫螢光 (direct immunofluorescence) 下曾報告有血管周圍 C3,以及較不一致的 IgM 與纖維蛋白原 (fibrinogen)。然而,如上所述,本病也可見白血球碎裂性血管炎 (leukocytoclastic vasculitis)。
Cryopin 相關週期性症候群 (Cryopin-associated Periodic Syndrome)(Muckle-Wells 症候群、家族性冷誘發自體發炎症候群與新生兒期發作多系統發炎疾病)
- Cryopin 相關週期性症候群 (Cryopin-associated periodic syndrome) 是一種具有可變外顯率 (variable penetrance) 的體染色體遺傳疾病,涵蓋一系列疾病譜,包括 Muckle-Wells 症候群 (Muckle-Wells syndrome)、家族性冷誘發自體發炎症候群 (familial cold autoinflammatory syndrome) 與新生兒期發作多系統發炎疾病 (neonatal-onset multisystem inflammatory disorder)。
- 所有這些病況的共同特徵為反覆發燒、關節痛與蕁麻疹 (urticaria)。它們可能不一地出現全身性類澱粉沉積症 (systemic amyloidosis)、失聰 (deafness)、結膜炎 (conjunctivitis) 與嚴重的神經學表現。
- 在此疾病譜中,familial cold autoinflammatory syndrome 代表輕度端,neonatal-onset multisystem inflammatory disorder 代表重度端。在 familial cold autoinflammatory syndrome 中,無失聰,且蕁麻疹的發作由寒冷所誘發。Amyloidosis 在 Muckle-Wells syndrome 中較常見,而嚴重的神經學表現則在 neonatal-onset multisystem inflammatory disorder 中較常見。
- 相同的血清前驅蛋白(serum amyloid A)產生 amyloid。Muckle-Wells syndrome 通常不會見到皮膚類澱粉沉積症 (cutaneous amyloidosis)。
- 本病與 NLRP3(又稱 CIAS1)的功能獲得性突變 (gain in function mutation) 有關,該基因編碼 cryopyrin,一種透過 caspase-1–interleukin-1 軸在發炎與細胞凋亡 (apoptosis) 調節中發揮作用的蛋白質。
- 曾有六名 Muckle-Wells syndrome 病人被描述在四肢與腹部出現硬化性、色素過度沉著的斑塊,並伴有多毛症 (hypertrichosis)。
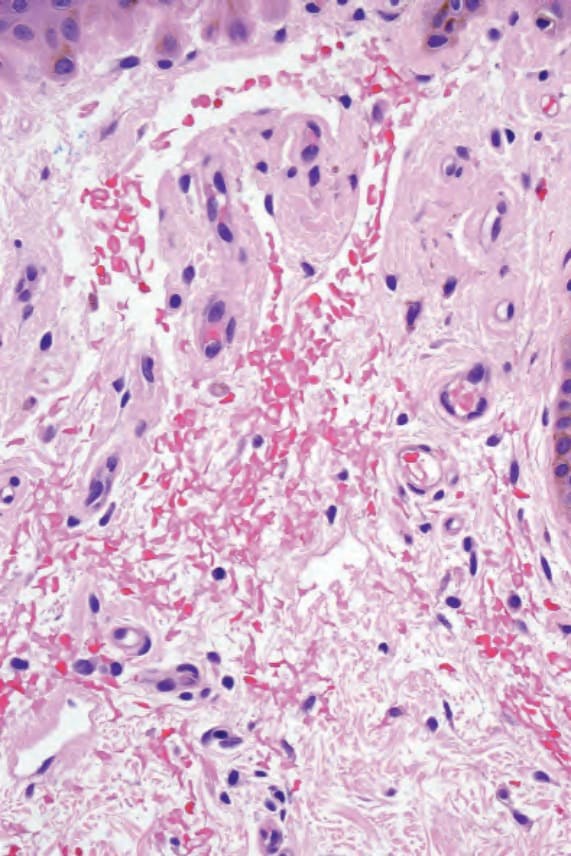
圖 13-48:原發性全身性類澱粉沉積症 (primary systemic amyloidosis):圖 13.47 的高倍視野。注意紅血球外滲 (red cell extravasation)。
Fig. 13.48 Primary systemic amyloidosis: high-power view of Fig. 13.47. Note the red cell extravasation.