12-Heredofamilial amyloidoses
12-Heredofamilial amyloidoses
Familial Mediterranean fever Clinical features This is an autosomal recessive inherited autoinflammatory disease. It is divided into two phenotypes: types 1 and 2. Type 1 is associated characterized by episodes of fever, serositis, peritonitis, synovitis, and in rare instances pericarditis and meningitis. Amyloidosis can result from the recurrent inflammatory episodes. In type 2, patients present initially with amyloidosis and are otherwise asymptomatic.1–5 Cutaneous lesions are rare and consist of Henoch-Schönlein purpura and erythema of the lower limbs mimicking erysipelas.1,6 Panniculitis, recurrent urticaria, polyarteritis nodosa, psoriasis-like lesions, bullous skin lesions, perivascular lymphocytic dermatitis, and sarcoidosis may also occur.7–14 Nail fold capillary abnormalities consisting of increased tortuosity and enlargement of capillary loops have also been documented.15,16 Cutaneous amyloid deposition has not been described.
Pathogenesis and histologic features Familial Mediterranean fever is cause by mutations in MEFV on chromosome 16p13, a gene that encodes pyrin, a protein involved in deactivating the immune response.5 Defects in pyrin lead to over production of interleukin-1, resulting in a proinflammatory state contributing to the formation of amyloid.17 In familial Mediterranean fever, a serum precursor protein forms the amyloid in this condition. This precursor is a HDL known as serum amyloid A.
good sensitivity and specificity (~70–90%), but others have demonstrated poor sensitivity in abdominal fat pad biopsies.10
Although frank clinical lesions are not commonly a feature of secondary amyloidosis, sometimes small deposits are found in specimens of normal skin.11 Usually these are present in a perivascular location, but may occasionally be present elsewhere in the dermis or even in subcutaneous fat.12 Deposition of amyloid around sweat glands may also be seen. Deposits are said to be focal and abdominal subcutaneous fat has been recommended as the site that is most likely to be positive.1,5,13 Hemodialysis-associated amyloidosis is a distinctive form of secondary amyloidosis and is described below.
The erysipelas-like lesions are characterized by a perivascular mixed infiltrate of lymphocytes, histiocytes, and neutrophils with leukocytoclasia.6 Vasculitis is not seen, although on direct immunofluorescence perivascular C3 and, less consistently IgM and fibrinogen, have been reported.6 However, as noted above, leukocytoclastic vasculitis may be seen in this disease.
Cryopin-associated periodic syndrome (Muckle-Wells syndrome, familial cold autoinflammatory syndrome and neonatal-onset multisystem inflammatory disorder) Cryopin-associated periodic syndrome is an autosomal inherited disease with variable penetrance that encompasses a spectrum of diseases that includes Muckle-Wells syndrome, familial cold autoinflammatory syndrome, and neonatal-onset multisystem inflammatory disorder.1,2 All of the conditions have in common recurrent fevers, joint pain, and urticaria. They variably may have systemic amyloidosis, deafness, conjunctivitis, and severe neurological manifestations.3,4 In the spectrum of disease, familial cold autoinflammatory syndrome represents the mild end of the spectrum and neonatal-onset multisystem inflammatory disorder the severe end.1 In familial cold autoinflammatory syndrome, there is no deafness and the episodes of urticaria are precipitated by cold. Amyloidosis is more common in Muckle-Wells syndrome and severe neurologic manifestations are more common in neonatal-onset multisystem inflammatory disorder.1,2 The same serum precursor protein (serum amyloid A) produces the amyloid. Cutaneous amyloidosis is not typically seen in Muckle-Wells syndrome. The disease is related to a gain in function mutation of NLRP3 (also called CIAS1) that encodes cryopyrin, a protein that plays a role in the regulation of inflammation, and apoptosis via caspase-1-interleukin-1 axis.1,2,5,6 Six patients with Muckle-Wells syndrome were described as having sclerotic, hyperpigmented plaques with hypertrichosis on the extremities and abdomen.7
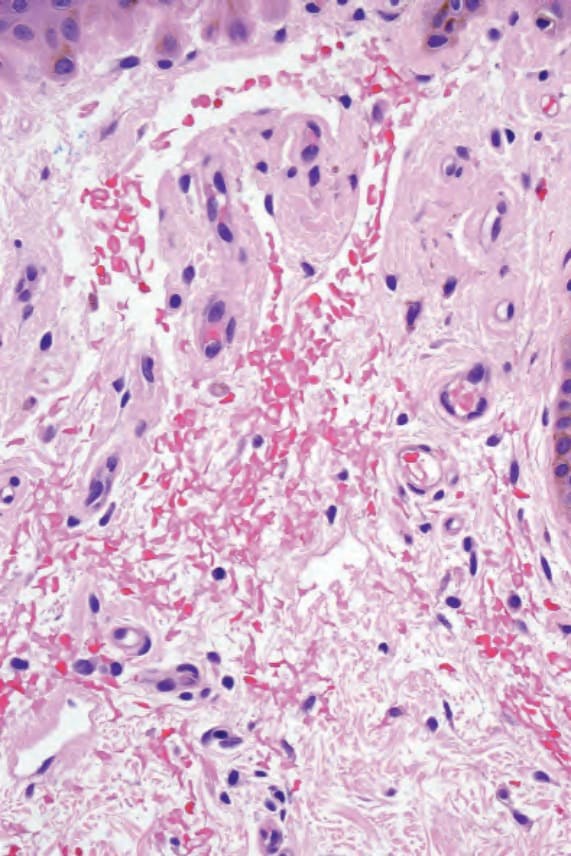
Fig. 13.48 Primary systemic amyloidosis: high-power view of Fig. 13.47. Note the red cell extravasation.