疾病定義與分類
- 表皮分解性水疱症 (epidermolysis bullosa, EB) 是指一群異質性疾病 (heterogeneous group),其皮膚(有時包括黏膜)在輕微外傷下即容易起水疱,因此有時亦使用「機械性水疱性疾病 (mechanobullous disease)」此一別名。
- 「epidermolysis(表皮分解)」這個描述性術語其實略不合邏輯,因為在數類 EB 中,表皮破壞 (epidermal disruption) 並非主要變化。儘管如此,自 Koebner 於 1886 年首次使用以來,epidermolysis bullosa 這個名稱在文獻中已根深柢固,仍是公認的術語。
- 所有型式的 EB 皆罕見;估計全球約有 500 000 人患有 EB。
EB 的分類
- 歷史上,EB 的分類一直困難重重,傳統上使用的大量名稱與人名命名 (eponyms) 更使其雪上加霜。
- 雖然這些早期觀察對於確立 EB 為一獨立疾病實體很重要,但 1960 年代 Pearson 邁出了重大的一步:他利用穿透式電子顯微鏡 (transmission electron microscopy) 顯示,皮膚組織裂開(水疱形成)的超微結構層次在 EB 的三大主要族群中各具特色:
- EB simplex 為表皮內 (intraepidermal)。
- junctional EB 為通過 lamina lucida。
- dystrophic EB 為 lamina densa 之下。
- (其後又另外加入第四類 Kindler syndrome,其水疱發生於可變的混合層次。)
- 事實上,這些不同的裂開層次至今仍構成目前 EB 分類的基礎(表 4-1)。
- 自 Pearson 最初的觀察以來,EB simplex 的概念與內容也隨著加入更多表淺性皮膚脆弱性疾病而擴大,包括胞橋小體 (desmosomes) 與角化 (cornification) 的遺傳性疾病(表 4-2)。
- 同樣地,junctional 與 dystrophic 型 EB 的分類也持續演進,不過其諸多變異型多依臨床特徵來界定(表 4-3 與表 4-4)。
- 雖然目前被視為 EB 型式的臨床病理實體已擴大,但大多數主要型式的 EB 發生於真皮–表皮交界 (dermal–epidermal junction) 或其鄰近,而這些型式 EB 所牽涉的關鍵蛋白與基因示意圖見圖 4-9。
超微結構、免疫組織化學與分子分類的演進
- 除了確立 EB 不同亞型中特定的水疱形成層次外,超微結構研究也能辨識出獨特的形態學異常,例如 EB simplex 中的角蛋白絲破壞 (keratin filament disruption)、junctional EB 中發育不良的半胞橋小體 (poorly formed hemidesmosomes),以及 dystrophic EB 中發育不全的固著纖維 (rudimentary anchoring fibrils)。
- 1980 年代,以基底膜帶抗體對 EB 皮膚進行免疫組織化學標誌成為有用的診斷補充,例如顯示某些型式 junctional EB 中 laminin-332 的免疫染色減少,以及 recessive dystrophic EB 中 type VII collagen 的免疫染色減少。
- 接著在 1990 年代,候選基因與致病性突變的發現,例如 EB simplex 中 KRT14 與 KRT5 的突變(keratins 14 與 5),以及 dystrophic EB 中 COL7A1 的突變(type VII collagen),揭開了分子診斷學 (molecular diagnostics) 的時代。
- 反映電子顯微鏡、免疫組織化學與分子層次上的這些進展,EB 的分類多年來持續演進,最近一份國際共識報告(發表於 2014 年)建議放棄大多數歷史性人名命名,並採用「洋蔥皮 (onion skin)」式的疾病命名法——不同的層次對應於可用的診斷評估方法:臨床表型、遺傳模式、皮膚的裂開層次、免疫組織化學、突變偵測等。
- 例如,一名表皮內水疱形成、水疱侷限於手掌與足底、且家族史符合體染色體顯性遺傳 (autosomal dominant transmission) 的患者,最初會被分類為 localized EB simplex。一旦完成突變確認,使用洋蔥皮法的最終診斷將為:EB simplex, localized, KRT5 mutation(錯義突變,missense mutation)。
- 一般認為此種診斷 EB 的格式在分類 EB 上兼具實用與學術價值,不過隨著更多發現出現,未來幾年很可能還會進一步修訂。
分子診斷與基因
- 分子研究目前已在 EB 異質性臨床亞型中辨識出 18 個不同基因的致病性突變(表 4-5)。
- 就診斷 EB 而言,患者 DNA 的定序(無論是 Sanger 定序,或使用全外顯子定序 (whole exome sequencing) 或選定基因套組 (selected gene panels) 的次世代定序),正逐漸成為主要的檢查方式,而皮膚切片的角色則略為下降。
- 此情形對於體染色體顯性型式的 EB simplex 或 dominant dystrophic EB 確實如此——這些型式的皮膚病理(包括超微結構與免疫組織化學)可能與正常皮膚無顯著差異,尤其當切片未包含新鮮水疱時。
- 儘管如此,皮膚切片仍是快速診斷體染色體隱性型式 EB 的有用方法,對於皮膚脆弱的新生兒特別有用,因為及早診斷對受影響嬰兒及其家庭的最佳臨床處置具有重要意義。有關診斷 EB 最適當的皮膚切片技術與檢查的細節,詳見第 2 章。
診斷方法與技術考量
- 多數用於診斷 EB 的抗體靶向跨膜蛋白 (transmembranous proteins),因此在石蠟包埋皮膚切片上效果不佳(或完全無效)。
- 就 EB 診斷而言,皮膚免疫組織化學最好在冷凍切片 (frozen sections) 上進行(見第 2 章)。
- 部分診斷性皮膚切片常會固定並處理以供穿透式電子顯微鏡檢查,不過通常只在皮膚免疫標誌無法確立診斷時才切片。
- 因此,過去十年間,隨著皮膚基底膜與上皮蛋白抗體的效用日增,需要穿透式電子顯微鏡來確立診斷的 EB 皮膚切片數量已減少超過 50%。隨著分子診斷學更為普及、更便宜且更快速,此數字很可能進一步下降。
- 免疫電子顯微鏡 (immunoelectron microscopy) 不用於常規 EB 診斷,主要用於研究,包括評估細胞與基因療法臨床試驗時的轉譯研究 (translational research)。
EB 主要亞型概要(依皮膚裂開層次)
- 表皮內 (Intraepidermal) — EB simplex:
- Suprabasal EB simplex:缺陷蛋白為 transglutaminase-5;plakophilin-1;desmoplakin;plakoglobin。
- Basal EB simplex:缺陷蛋白為 keratins 5 與 14;plectin;exophilin-5;230-kDa bullous pemphigoid antigen/dystonin。
- lamina lucida 內 (Intralamina lucida) — Junctional EB:
- Junctional EB generalized:laminin-332;type XVII collagen;α6、β4、α3 integrin 次單元。
- Junctional EB localized:type XVII collagen;laminin-332;α6、β4 integrin 次單元。
- lamina densa 之下 (Sublamina densa) — Dystrophic EB:
- Dominant dystrophic EB:type VII collagen。
- Recessive dystrophic EB:type VII collagen。
- 混合型 (Mixed) — Kindler syndrome:kindlin-1(Fermitin family homologue-1)。
- 註:所有型式與亞型的 dystrophic EB,其靶向蛋白皆為 type VII collagen。
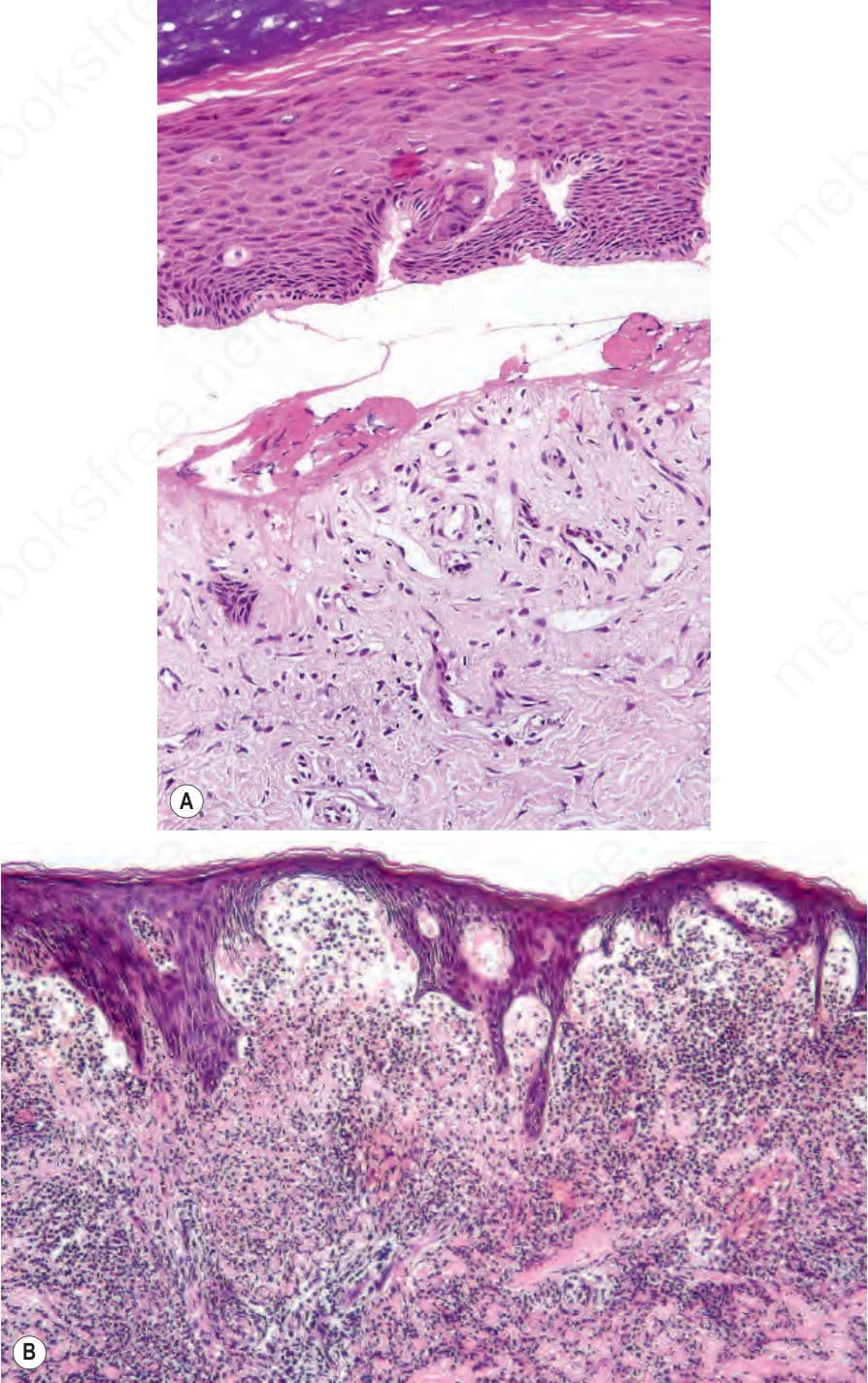
圖 4-1:表皮下水疱的分類:病灶可再細分為 (A) 細胞稀少型 (cell-poor) 與 (B) 細胞豐富型 (cell-rich) 變異。
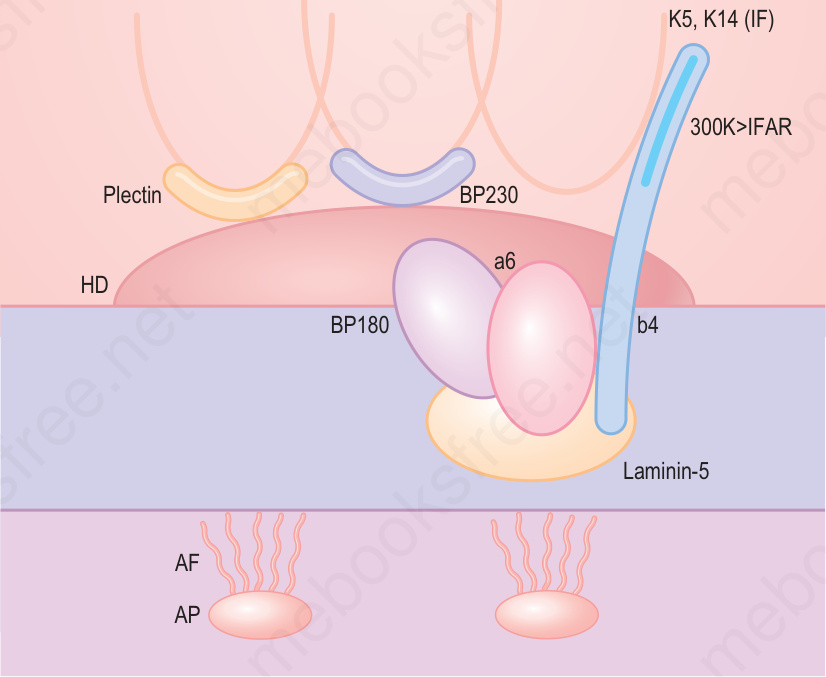
圖 4-2:基底膜組成成分:水疱可分類為發生於 lamina lucida (LL) 內者,以及發生於 lamina densa (LD) 之下者。(AF, anchoring fibrils;AP, anchoring plaque;CM, cell membrane。)
圖 4-3:分裂皮膚免疫螢光 (split skin immunofluorescence)。
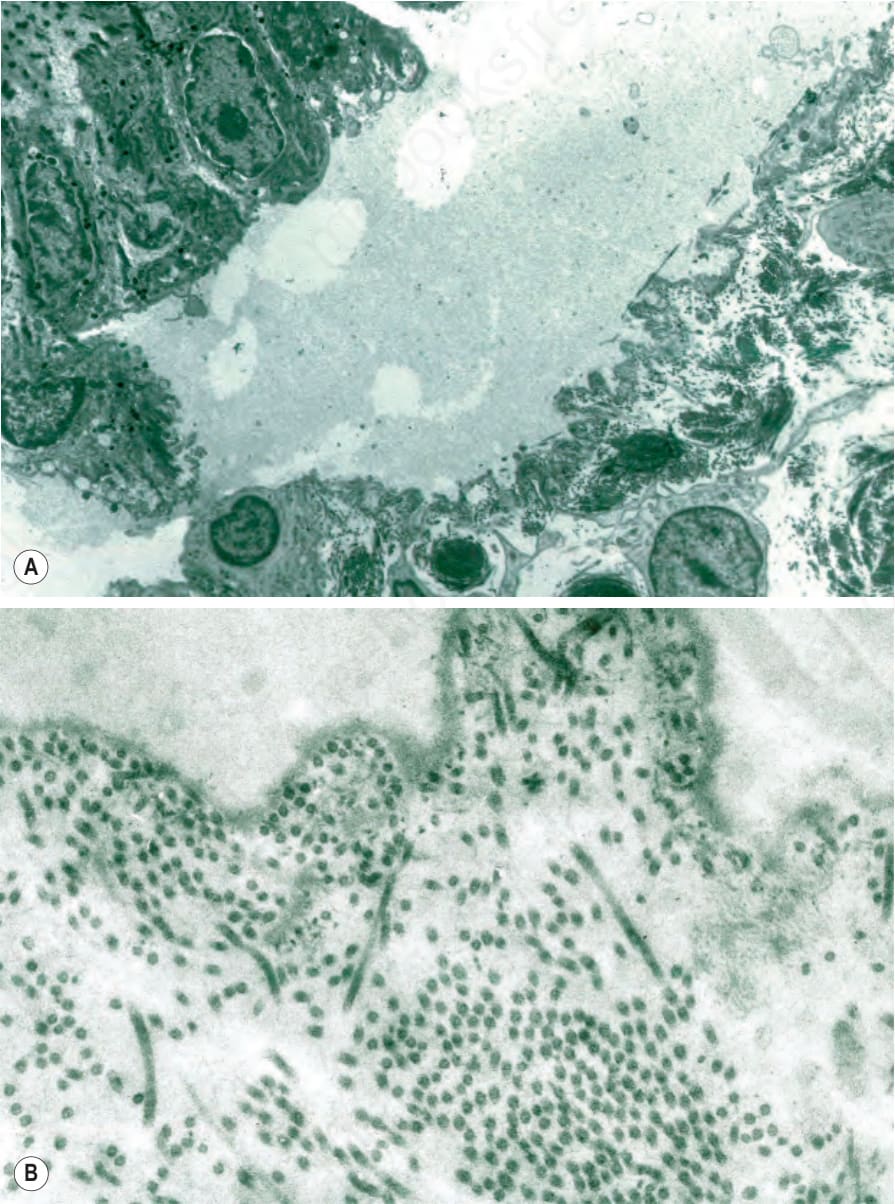
圖 4-4:(A, B) 分裂皮膚免疫螢光:分裂通過 lamina lucida,lamina densa 襯於人工水疱腔的底部。
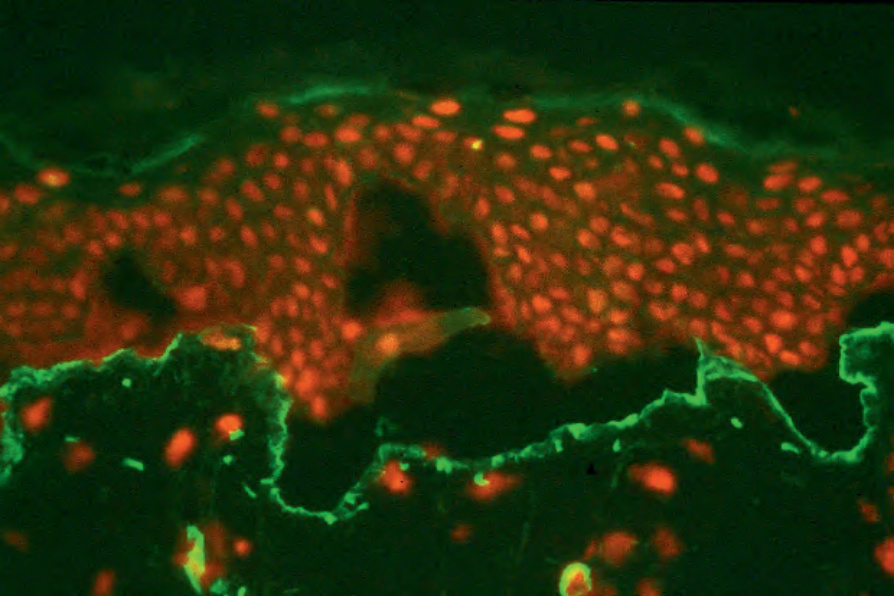
圖 4-5:分裂皮膚免疫螢光:type IV collagen 襯於分裂皮膚人工水疱的底部,故水疱形成於 lamina lucida 之內。承蒙英國倫敦 Institute of Dermatology 之 B. Bhogal, FIMLS 提供。
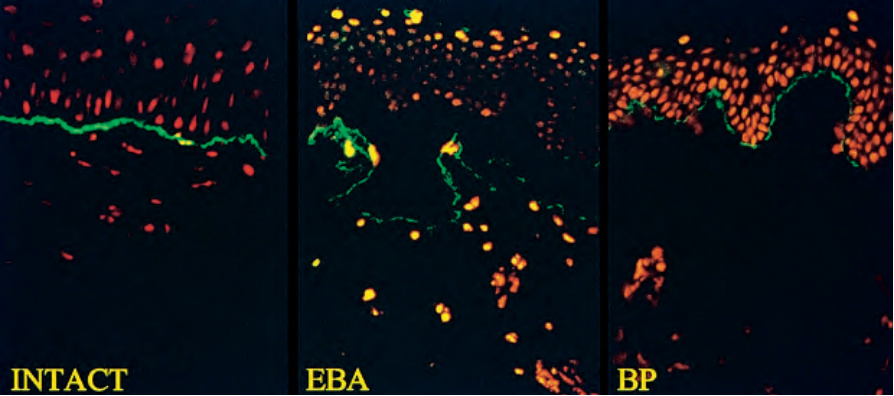
圖 4-6:分裂皮膚免疫螢光:(左)基底膜處的線狀 IgG;(中)在 epidermolysis bullosa acquisita (EBA) 中,抗體結合於水疱腔底部;(右)在 bullous pemphigoid (BP) 中,抗體結合於水疱頂部。承蒙英國倫敦 Institute of Dermatology 之 B. Bhogal, FIMLS 提供。
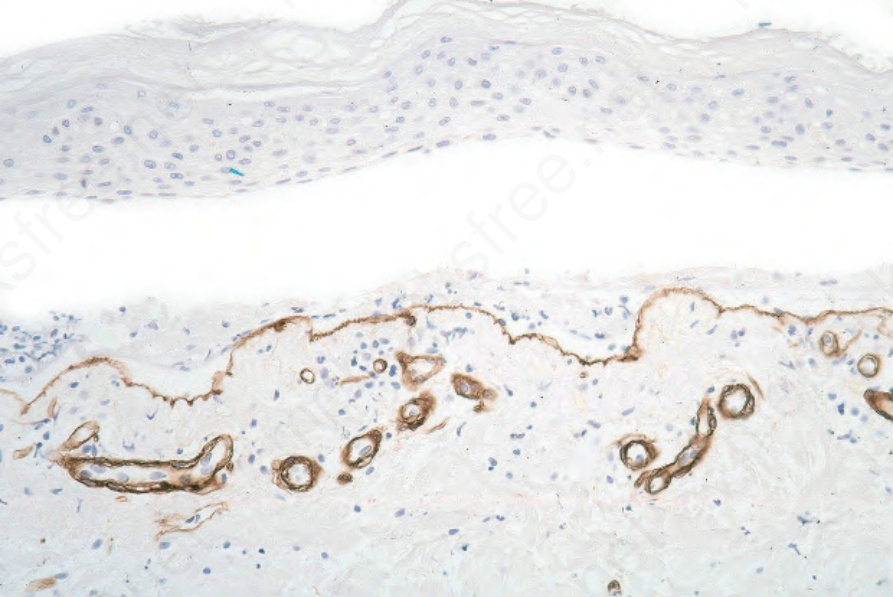
圖 4-7:石蠟包埋免疫過氧化酶抗原定位:在 bullous pemphigoid 中,type IV collagen 出現於水疱底部。
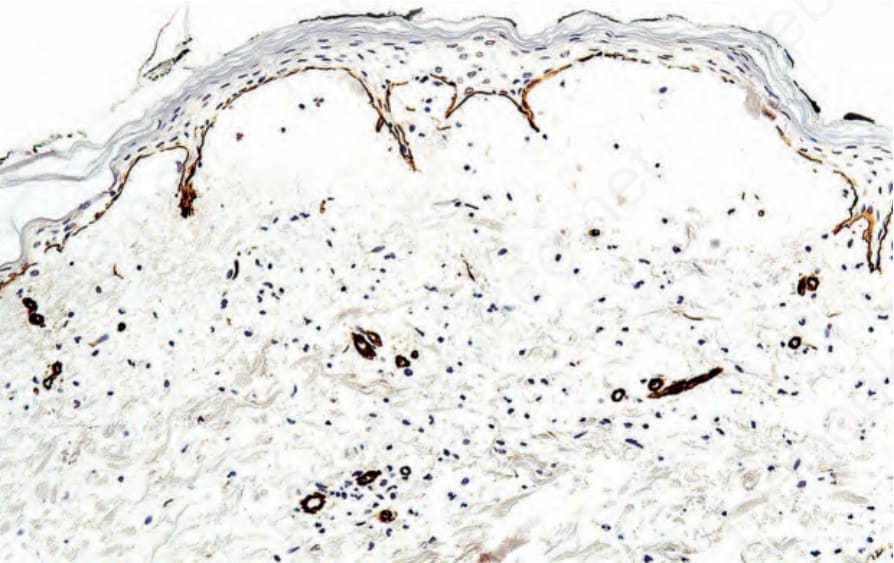
圖 4-8:石蠟包埋免疫過氧化酶抗原定位:在 epidermolysis bullosa acquisita 中,type IV collagen 出現於水疱腔頂部。
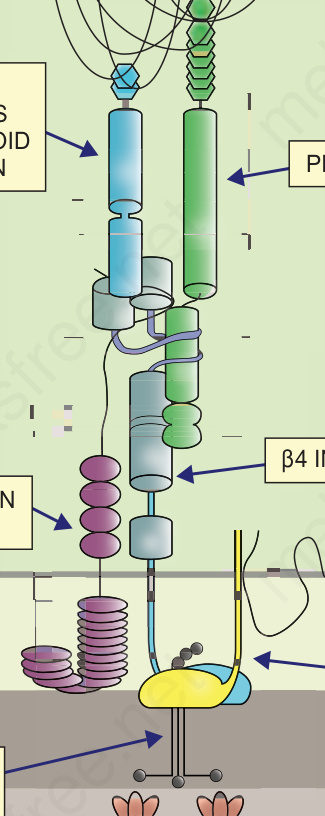
圖 4-9:真皮–表皮交界處半胞橋小體黏附複合體 (hemidesmosome adhesion complexes) 內主要黏附蛋白的示意圖,以及它們在不同型式 EB 中的牽涉。(AD = autosomal dominant;AR = autosomal recessive。)
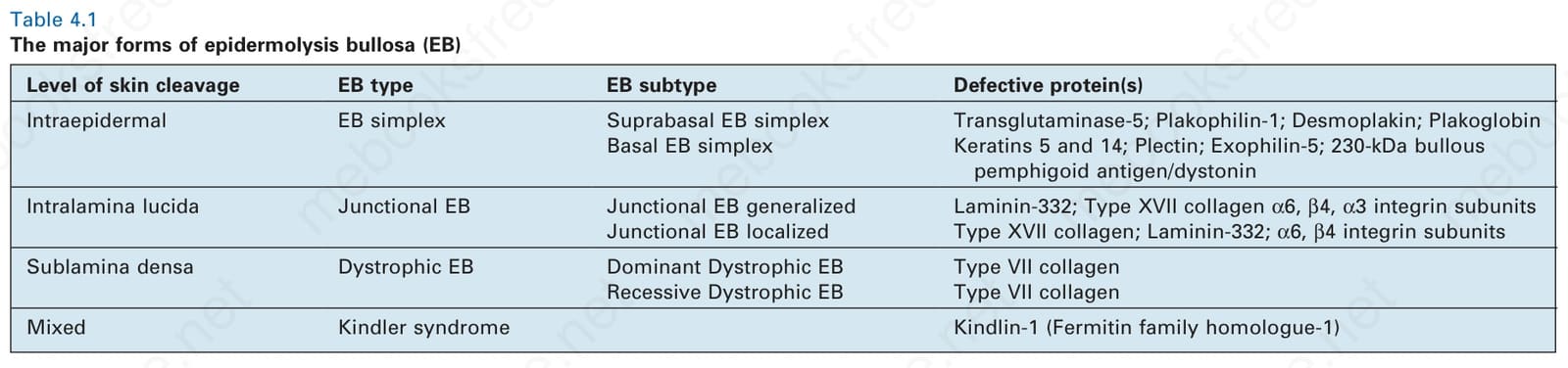
表 4-1:表皮分解性水疱症 (epidermolysis bullosa, EB) 的主要型式。
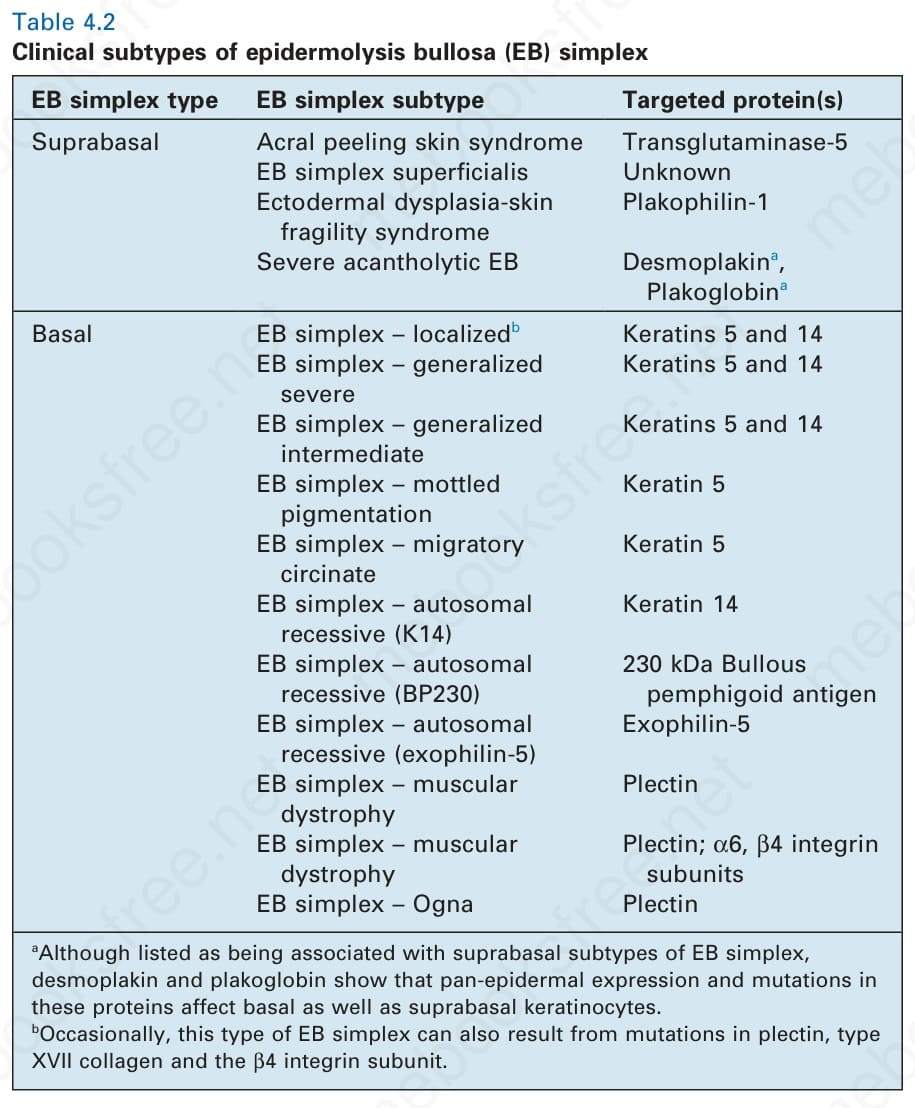
表 4-2:表皮分解性水疱症單純型 (epidermolysis bullosa simplex) 的臨床亞型。
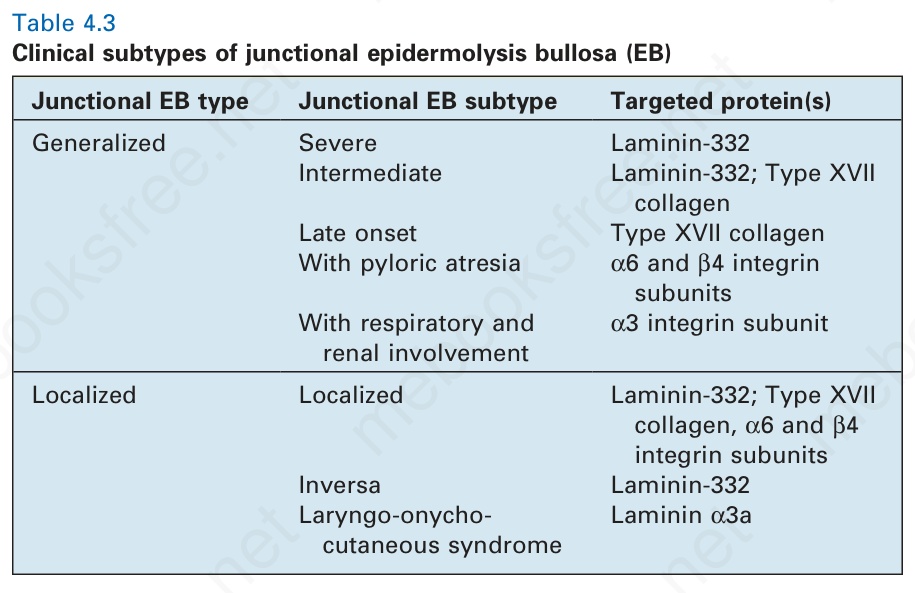
表 4-3:交界型表皮分解性水疱症 (junctional epidermolysis bullosa) 的臨床亞型。
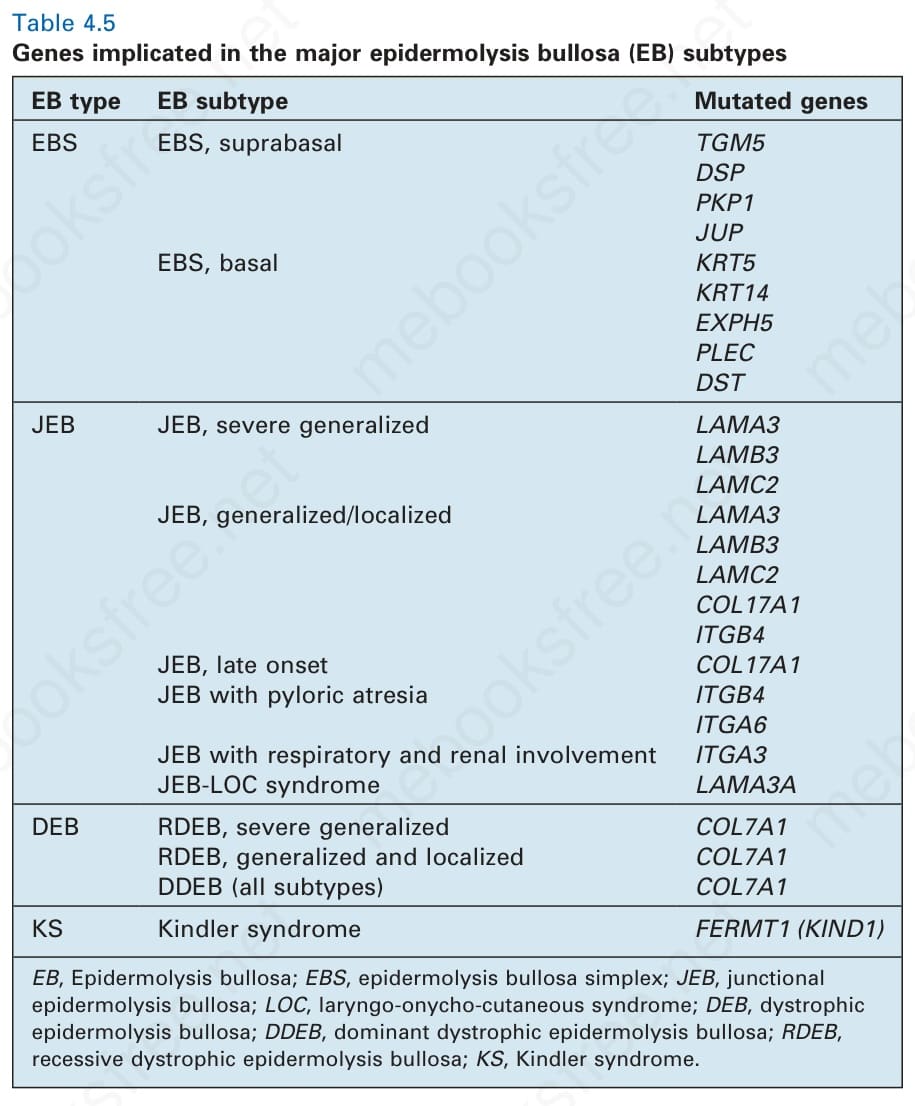
表 4-5:主要表皮分解性水疱症 (EB) 亞型所牽涉的基因。
診斷策略與材料考量
- 對皮膚病理醫師而言,一個關鍵問題始終是:EB 最好依皮膚/分子病理來分類,還是依臨床形態來分類;因此在以下各節中,兩種選項皆會呈現。
- 另一個重要問題是:如何以可取得的材料盡可能妥善地診斷 EB。
- 對於以福馬林固定並包埋於石蠟的皮膚,能否成功診斷可能取決於是否有新鮮水疱 (fresh blister)。
- 多數臨床醫師都知道,切取已存在超過 12 小時的水疱,很可能因再上皮化 (re-epithelialization) 而使病理醫師難以判定真正的裂開平面,因此較偏好取樣經摩擦但未起水疱的皮膚 (rubbed non-blistered skin)(見第 2 章)。
- 抗原定位 (antigen mapping) 與免疫過氧化酶染色 (immunoperoxidase staining) 可使用 type IV collagen 抗體進行,藉此判定 lamina densa 位於水疱的頂部(dystrophic EB)或底部(EB simplex 與 junctional EB)。
- 也可使用選擇性探針來作免疫診斷,例如 type VII collagen(在 recessive dystrophic EB 中減少)。
- 儘管如此,多數用於診斷 EB 的抗體靶向跨膜蛋白 (transmembranous proteins)。
EB 的臨床特徵
EB simplex
- EB simplex 的各亞型再細分為基底型 (basal) 與基底上型 (suprabasal) 變異,表現為至少 15 種不同的臨床疾病(表 4-2)。
Localized EB simplex(侷限型)
- 這是最常見的 EB 型式。遺傳為體染色體顯性 (autosomal dominant)。
- 主要侵犯足底與手掌(圖 4-10)。此病在溫暖天氣下通常較嚴重。
- 足部多汗症 (hyperhidrosis) 常見;此會增加摩擦,亦使起水疱惡化。
- 水疱通常癒合而不留疤痕,也不形成粟粒疹 (milia)。胼胝 (calluses) 非常常見,尤其在成人。
- 口腔起水疱發生於少於 25% 的病例。毛髮與牙齒正常。
Acral peeling skin syndrome(肢端剝脫性皮膚症候群)
- localized 體染色體顯性 EB simplex 的一個鑑別診斷是體染色體隱性 (autosomal recessive) 的肢端剝脫性皮膚症候群,雖然兩者皆歸類為 EB simplex 的型式。
- 水疱通常發生於足部,但常於足趾的側面或背側更為明顯(圖 4-11)。
- 起水疱層次位於顆粒層 (granular layer) 之上,但由於肢端皮膚的角質層 (stratum corneum) 較厚,臨床後果可能與通過基底角質細胞層起水疱者幾乎相同。
Generalized severe EB simplex(泛發重症型)
- EB simplex 另一個常見亞型為泛發重症變異,先前稱為 Dowling-Meara EB simplex。遺傳為體染色體顯性。
- 水疱傾向成群出現,因此早期使用 EB herpetiformis(疱疹樣 EB)一詞(圖 4-12)。
- 嬰兒期起水疱可能嚴重,伴有黏膜侵犯、指甲脫落,以及粟粒疹 (milia) 形成。
- 其獨特特徵是軀幹、四肢與頸部出現自發性疱疹樣 (herpetiform)、環狀 (annular) 或弧形 (arcuate) 的水疱。手掌與足底常出現不規則過度角化 (hyperkeratosis) 或角皮症 (keratoderma)。整體狀況傾向隨年齡改善。
Generalized intermediate EB simplex(泛發中度型)
- 此 EB 亞型包含先前稱為 Koebner EB simplex 的遺傳性起水疱。多數病例為體染色體顯性。
- 雖然通常輕微,但約 60% 患者有侷限性疤痕 (localized scarring),約 15% 有粟粒疹 (milia)。毛髮、牙齒與指甲的發育正常。
- 水疱出現於出生後第一年內,也可能出生時即存在。嬰兒期常出現於枕部 (occiput)、背部與腿部,兒童期則常侵犯手與足,但手掌與足底不像 localized EB simplex 那樣優先受侵犯。
- 與其他型式 EB simplex 相同,起水疱在溫暖天氣下較嚴重。
EB simplex autosomal recessive keratin 14(體染色體隱性 keratin 14 型)
- 雖然遠較體染色體顯性疾病少見,但 keratin 14 的隱性突變病例可能類似 dominant generalized intermediate EB simplex。
- 水疱常散在分布,通常伴有輕微的掌蹠角皮症 (palmoplantar keratoderma),以及不同程度的指甲營養不良 (nail dystrophy)、萎縮性疤痕 (atrophic scarring)、色素沉著 (hyperpigmentation),以及口腔與生殖器起水疱。
- 表皮中 keratin 14 表現常完全喪失,不過其表型通常較某些顯性病例為輕。目前尚未有牽涉 keratin 5 的隱性病例報告。
EB simplex with muscular dystrophy(合併肌肉失養症型)
- 遺傳性皮膚脆弱可與肌肉失養症 (muscular dystrophy) 並存,不過也曾描述合併重症肌無力 (myasthenia gravis) 與脊髓性肌肉萎縮症 (spinal muscular atrophy)。
- 水疱侵犯皮膚與口腔黏膜,於出生時或出生後不久出現。
- 肌肉無力與萎縮可能嚴重並於幼兒期顯現,或較輕微而僅在較晚的生命期才被偵測到。
- 起水疱可能廣泛,但也可侷限於手與足。可有萎縮性疤痕、粟粒疹、指甲營養不良與禿髮 (alopecia)。
- 聲門上疤痕 (supraglottic scarring) 與聲音嘶啞可能需要氣管切開術 (tracheostomy)。此病為體染色體隱性,源於 plectin 的突變。
EB simplex with pyloric atresia(合併幽門閉鎖型)
- 幽門閉鎖 (pyloric atresia) 罕見地可發生於 EB simplex 的情境中——其多數病例被歸類為 junctional 型 EB。
- 受影響嬰兒傾向有泛發性皮膚疾病。雖然目前僅有少數病例報告,此實體似乎與合併幽門閉鎖的 junctional EB 一樣嚴重,臨床上不可能與之區分。
- 有廣泛的水疱與糜爛,增加感染風險,於新生兒期早期即死亡。遺傳為體染色體隱性,牽涉 plectin 的突變。
EB simplex autosomal recessive BP230(體染色體隱性 BP230 型)
- 導致 230-kDa bullous pemphigoid antigen 完全喪失的突變,造成一種罕見的體染色體隱性 EB simplex。
- 起水疱終生持續且泛發,但臨床上輕微,僅有少數主要為肢端的水疱,可擴大至數公分,但屬非炎症性,癒合後不留疤痕,僅有輕微的發炎後色素異常 (postinflammatory pigmentary anomalies)。口腔與生殖器黏膜不受侵犯。
EB simplex with mottled pigmentation(合併斑駁色素沉著型)
- 與其他型式 EB simplex 的區別在於有色素變化,於出生時即存在或在嬰兒期出現。
- 呈網狀 (reticulate) 分布的小型、棕褐色 (tan-colored) 斑疹病灶,可從肢端部位蔓延至軀幹,並隨年齡淡化(圖 4-13)。
- 它們可遍布整個皮膚表面,但優先侵犯頸部、上軀幹或四肢。起水疱可為侷限性,或變得較為泛發。
- 多數病例源於 keratin 5 中一個特定的異型合子錯義突變 (heterozygous missense mutation)。
EB simplex autosomal recessive exophilin-5(體染色體隱性 exophilin-5 型)
- EXPH5(編碼 exophilin-5,亦稱 Slac2-b)的體染色體隱性功能喪失突變 (loss-of-function mutations),造成輕微、散在、由外傷誘發的皮膚脆弱,不過出生後不久起水疱可為泛發。
- 然而在較晚的嬰兒期,臨床特徵多為四肢的結痂糜爛 (crusted erosions),少有完整水疱。未見黏膜異常。
- 典型上,疾病嚴重度隨年齡改善,甚至可能完全緩解。
EB simplex plakophilin-1 deficiency(plakophilin-1 缺乏型)
- 此疾病為第一個被描述的胞橋小體 (desmosomes) 遺傳性疾病;亦稱為外胚層發育不良–皮膚脆弱症候群 (ectodermal dysplasia skin fragility syndrome)。遺傳為體染色體隱性。
- 出生時即有廣泛糜爛,伴有明顯的口周龜裂 (perioral cracking) 與毛髮稀少 (hypotrichosis)。
- 嬰兒期皮膚表現演變為外傷誘發的鱗屑結痂、明顯的掌蹠角皮症伴疼痛性裂隙,以及指甲營養不良。
- 多數病例有顯著的頭皮毛髮與眉毛脫落,口周變化持續存在。
- 與某些其他遺傳性胞橋小體疾病不同,此病無心臟侵犯,因為 plakophilin-1 不在心臟中表現。
EB simplex Ogna(Ogna 型)
- 此體染色體顯性疾病以挪威首個受影響家族的發源村莊命名。
- 有季節性的手足起水疱,偶爾發生於其他部位。
- 此罕見 EB 亞型在臨床上可藉由泛發的瘀傷傾向 (bruising tendency)、出血性大疱 (hemorrhagic bullae) 與趾甲營養不良 (toenail dystrophy) 來辨別。
- 此病源於 plectin 中一個特定的錯義突變,使該蛋白更易被蛋白水解 (proteolysis),從而降低其表現與功能。
EB simplex migratory circinate(遊走性環狀型)
- 這是一種罕見的體染色體顯性 EB simplex,特徵為遊走性環狀紅斑 (migratory circinate erythema) 與發炎後色素沉著 (post-inflammatory hyperpigmentation)。
- 出生時即存在,起水疱為泛發。
- 其根本原因是 keratin 5 中一個非典型突變,導致皮膚發炎並伴有 T 細胞浸潤 (T-cell infiltrate)。
EB simplex desmoplakin deficiency(desmoplakin 缺乏型)
- 胞橋小體斑塊蛋白 (desmosomal plaque protein) desmoplakin 的體染色體隱性突變,可造成一種非常嚴重的遺傳性皮膚脆弱。
- 原先稱為「lethal acantholytic EB(致死性棘層鬆解性 EB)」,不過鑑於「lethal(致死)」此類情緒性字眼對家庭可能造成的衝擊,以及某些病例疾病病程可能難以預測,最新的 EB 分類已試圖避免使用此類用語。
- 起水疱於出生時即存在且泛發。一個特徵是出現滲液性糜爛 (oozing erosions) 而非明顯水疱。
- 其他發現包括明顯異常的指甲、新生兒牙齒 (neonatal teeth)、口腔內糜爛與禿髮。胃腸道、泌尿生殖道與呼吸道亦可能受侵犯。
EB simplex plakoglobin deficiency(plakoglobin 缺乏型)
- plakoglobin(一種見於胞橋小體斑塊與黏附連結 (adherens junctions) 的蛋白)的體染色體隱性突變,亦可造成一種非常嚴重的遺傳性皮膚脆弱。
- 原先稱為「lethal congenital EB(致死性先天性 EB)」,不過如同 desmoplakin 的情況,最新的 EB 分類共識已避免使用「lethal」一詞,而傾向改用 skin fragility plakoglobin deficiency(或 severe acantholytic EB)。
- 臨床上有廣泛糜爛與大量經皮液體流失 (transcutaneous fluid loss),導致預後不良。某些遺傳性 plakoglobin 異常可能有心臟侵犯。
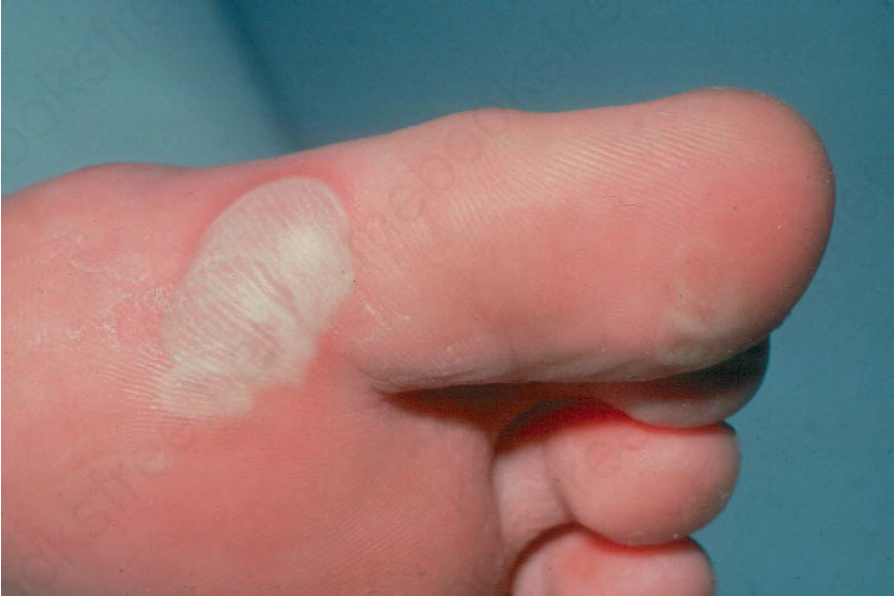
圖 4-10:Localized EB simplex:侵犯足趾的典型肢端病灶。其蒼白色澤是因水疱頂部明顯增厚所致。承蒙英國倫敦 St John’s Institute of Dermatology 提供。
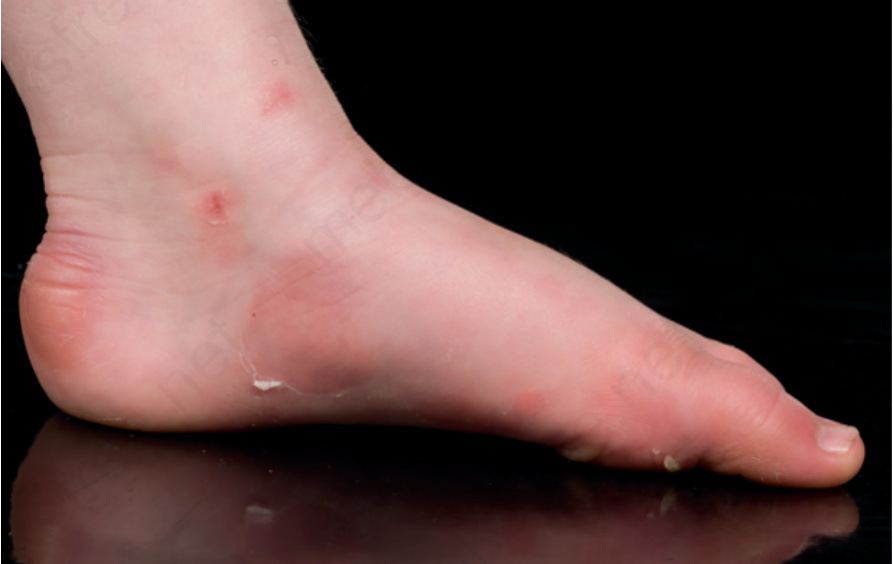
圖 4-11:Acral peeling skin syndrome:踝部與足背內側出現剝脫 (peeling) 與紅斑 (erythema) 的徵象,並延伸至足趾。
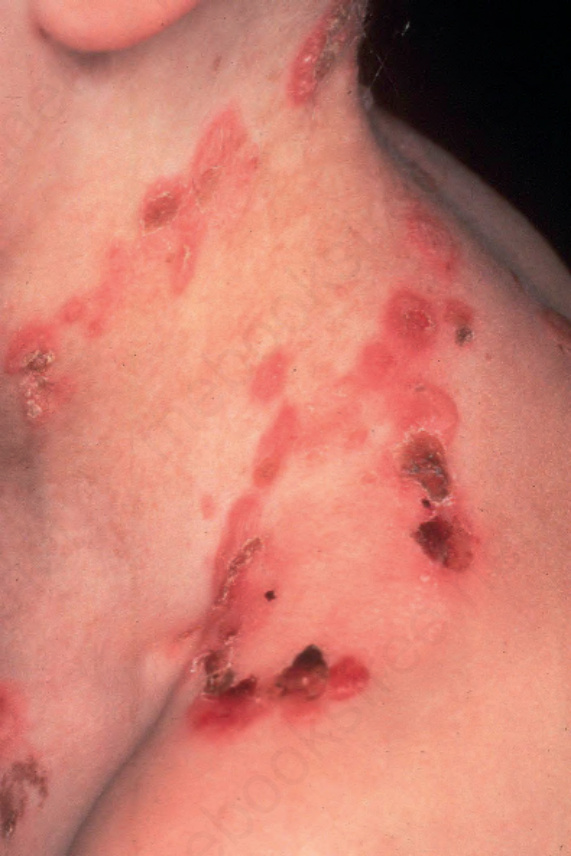
圖 4-12:Generalized severe EB simplex:顯示特徵性的水疱與糜爛成群分布。承蒙英國倫敦 St John’s Institute of Dermatology 之 R.A.J. Eady, MD 提供。
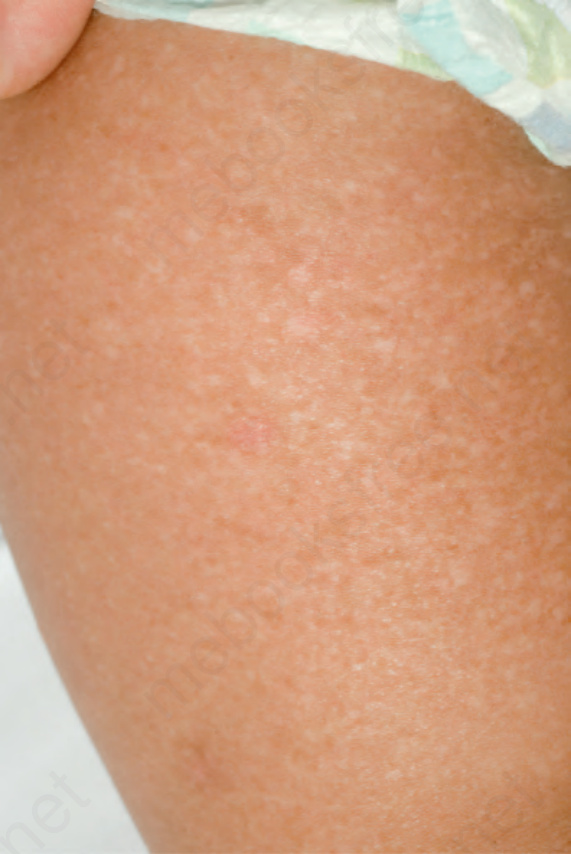
圖 4-13:EB simplex with mottled pigmentation:瀰漫性的色素增多與色素減退,伴少數小片紅斑與偶見的微小水疱。承蒙英國倫敦 St John’s Institute of Dermatology 之 J.E. Mellerio 提供。
單純型表皮鬆解水疱症之亞型續論 (EB Simplex Subtypes, continued)
淺表性單純型 EB (EB simplex superficialis)
此為一種體染色體顯性 (autosomal dominant) 疾病,其特徵為出現淺表性糜爛 (superficial erosions) 而非完整水疱,類似於落葉型天疱瘡 (pemphigus foliaceus) 所見者。表皮裂隙 (epidermal cleavage) 通常恰位於角質層 (stratum corneum) 正下方。癒合後會留下局部萎縮性疤痕 (atrophic scarring) 或發炎後色素沉著 (postinflammatory hyperpigmentation)。此 EB simplex 亞型的分子基礎仍不清楚,因此尚不確定此疾病是否真為一個獨立的臨床病理實體。
接合處型表皮鬆解水疱症 (Junctional EB)
幾乎所有 junctional EB 的臨床變異型皆以體染色體隱性 (autosomal recessive) 遺傳為特徵,且水疱形成於透明板 (lamina lucida) 的層面。傳統上,junctional EB 分為兩大類:泛發型 (generalized) 與局限型 (localized),每一類各有數種亞型(表 4.3)。在更新的 EB 分類中,Herlitz、non-Herlitz、generalized atrophic benign、cicatricial、progressive 與 atrophicans 等名詞已不再使用,「lethal」或「letalis」等字眼亦不再被視為適當的診斷標籤。
泛發重度接合處型 EB (Generalized severe junctional EB)
此為 junctional EB 中最嚴重的型態。水疱與糜爛於出生時或出生後不久即出現,並迅速變為泛發性。整片皮膚都很脆弱,移動嬰兒可能會造成大範圍的水疱形成(圖 4.14)。甲床 (nail bed) 受侵犯非常常見(圖 4.15)。糜爛區域常癒合得很慢,並可能導致萎縮性疤痕。一般不會見到粟粒疹 (milia)。口腔與咽部黏膜的侵犯很常見且可能很嚴重;聲音沙啞 (hoarseness) 與喘鳴 (stridor) 可能提示喉部或聲門上 (supraglottic) 的侵犯,尤其是可能危及生命的狹窄 (stenosis) 或狹窄性瘢痕 (stricture)。許多嬰兒在嬰兒期早期即因勢不可擋的感染或因生長遲滯 (failure to thrive) 而死亡,但那些撐過頭幾個月的嬰兒往往會發展出特徵性的病灶,表現為不癒合、結痂的糜爛,內含過度增生的肉芽組織 (exuberant granulation tissue)。
泛發中度接合處型 EB (Generalized intermediate junctional EB)
此 junctional EB 亞型的早期臨床病程可能與 generalized severe 變異型相似,但病人通常可存活至成年。水疱的嚴重度隨年齡漸減。黏膜會受侵犯,牙齒呈現嚴重的琺瑯質缺損 (enamel defects) 或可能無法正常萌發。指甲常呈失養性 (dystrophic) 或缺失。萎縮性疤痕為其特徵。亦有影響頭皮、眉毛與睫毛的禿髮 (alopecia),體毛也稀疏或缺失。大型色素性痣 (large pigmented nevi) 或後天獲得性斑狀色素過度沉著 (acquired macular hyperpigmented lesions) 為典型表現。在此型 junctional EB 中,常可觀察到不會起水疱的小片皮膚;此現象稱為回復性鑲嵌現象 (revertant mosaicism,亦稱為自然基因治療 natural gene therapy),代表突變基因其中一個複本發生了自發性的矯正。
泛發遲發型接合處型 EB (Generalized late-onset junctional EB)
此為一種罕見的體染色體隱性 junctional EB 亞型,與 generalized intermediate junctional EB 重疊,差別在於症狀的發作延遲——常直到兒童期才開始,典型為 5 至 8 歲之間。起初,外傷誘發的水疱主要發生於手與足,雖然可能先有指甲失養 (nail dystrophy)。之後,膝與肘亦受波及。進行性的萎縮變化導致指紋型態 (fingerprint patterns) 提早消失與輕度手指攣縮 (finger contractures)。牙齒琺瑯質可能有缺損,舌乳頭 (tongue papillae) 可能消失。口腔黏膜有時會受侵犯。
合併幽門閉鎖之接合處型 EB (Junctional EB with pyloric atresia)
水疱通常於出生時即存在,懷孕過程常合併羊水過多 (polyhydramnios)。病灶通常分布廣泛,可導致萎縮性疤痕(圖 4.16)。牙齒發育不全 (hypoplastic)、缺乏正常琺瑯質,指甲呈失養性。早期嘗試餵食會導致嘔吐。多數病例無法存活過嬰兒期早期。在那些存活下來的兒童中,其他特徵包括血尿 (haematuria)、排尿疼痛 (dysuria) 與反覆泌尿道感染。幽門管 (pyloric canal) 常完全閉塞(圖 4.17),需要手術矯正,雖然其潛在分子病理的性質通常會影響預後。
合併呼吸道與腎臟侵犯之接合處型 EB (Junctional EB with respiratory and renal involvement)
此為近期才加入 junctional EB 分類的一型。其特徵包含先天性腎病症候群 (congenital nephrotic syndrome)、間質性肺病 (interstitial lung disease) 與皮膚脆弱性,肇因於 α3 integrin 的體染色體隱性突變。腎臟與呼吸道的特徵在臨床上居主導地位,皮膚水疱可能只是次要特徵且僅較晚期才出現。口腔黏膜不受侵犯。頭皮毛髮、眉毛與睫毛細而稀疏,指甲可能呈失養性。預後不佳,會有反覆肺部感染與多器官衰竭,與已知 α3 integrin 在數個組織的分布一致。
局限型接合處型 EB (Localized junctional EB)
junctional EB 的局限型確實存在:典型臨床表現包括指甲失養、牙齒琺瑯質變化,以及僅侵犯小腿下段與足部的水疱形成。在某些個體中,與角化過度 (hyperkeratosis) 相關的慢性疼痛性糜爛會發生於足底,雖然尚不清楚為何小腿下段會是水疱的好發部位。某些病例的水疱於新生兒期即開始,而另一些則可能為遲發性疾病。
局限倒置型接合處型 EB (Localized junctional EB inversa)
在罕見的局限型 junctional EB 倒置 (inversa) 亞型的新生兒期,整片皮膚可能脆弱並伴泛發性水疱。然而到後期,病灶主要侵犯腹股溝、會陰與腋窩,故有「inversa」(倒置)之描述。癒合後可能留下小而萎縮的白色條紋。發育不良的牙齒 (dysplastic teeth)、角膜糜爛 (erosions of the cornea),以及足部與指甲失養都是其特徵。為何會偏好屈側部位 (flexural site) 受侵犯則不得而知。
局限型接合處型 EB 喉-甲-皮症候群 (Localized junctional EB laryngo-onycho-cutaneous syndrome)
此 junctional EB 亞型於嬰兒期開始,表現為侵犯面部(主要在鼻與口周圍)的慢性糜爛,雖然糜爛亦見於四肢、軀幹與生殖器。指甲亦受侵犯,伴有明顯的甲周 (periungual) 與甲下 (subungual) 發炎,而聲音沙啞為一普遍特徵。牙齒可能有缺刻 (notched)。有顯著的皮膚與黏膜肉芽組織,可導致傷口癒合延遲、喉部阻塞與失明。此疾病肇因於 laminin α3 多肽 (laminin α3 polypeptide) 一特定剪接變異型 (splice variant) 的體染色體隱性突變。
失養型表皮鬆解水疱症 (Dystrophic Epidermolysis Bullosa)
dystrophic EB 可為體染色體隱性或體染色體顯性遺傳(表 4.4)。臨床上,dystrophic EB 的特徵為皮膚脆弱、水疱形成、疤痕、指甲失養與粟粒疹形成。黏膜侵犯很常見,糜爛與疤痕可影響口、食道、生殖器與肛門。某些隱性與顯性 dystrophic EB 病例之間可能有臨床重疊,這會使遺傳諮詢變得困難,尤其在偶發 (sporadic) 病例。最新的 EB 分類不再包含任何以人名命名的亞型,並認知到表型外觀在病人之間差異甚大,且鑑於臨床外觀的廣泛光譜,在某些病例中,要將個體歸入單一類別的診斷標籤可能有些武斷。
泛發顯性失養型 EB (Dominant dystrophic EB generalized)
dominant dystrophic EB 的水疱主要發生於覆蓋骨性突起 (bony prominences) 之皮膚受外傷後,例如膝與踝,以及手或足的背側(圖 4.18)。最一致的發現為伴粟粒疹形成的局部疤痕與失養性指甲。指甲失養可能是此疾病最重要的診斷特徵,尤其在成人,因為許多病人只有有限的水疱與疤痕,且隨年齡而變得較不明顯。口腔內的水疱通常輕微,牙齒一般正常。然而,肛周病灶可能造成相當大的疼痛,尤其在兒童。如 Bart syndrome 等名詞已過時。曾被認為是 dominant dystrophic EB 亞型特異性病徵 (pathognomonic) 的皮膚特徵,如白色丘疹樣病灶 (albopapuloid lesions),現已認知會出現於數種型態的 dystrophic EB 中。
肢端型顯性失養型 EB (Dominant dystrophic EB acral)
此 dominant dystrophic EB 亞型的性質並未被精確界定,因為「acral」(肢端)一詞在已發表的文獻中並未廣泛使用。將其納入最新的 EB 分類,是為了協助描述那些皮膚侵犯型態較局限、通常侵犯手與足的 dominant dystrophic EB 病例(圖 4.19)。然而,之所以使用「acral」一詞而非「localized」,是因為儘管大部分皮膚相對未受侵犯,仍可能發生某些口腔或食道的侵犯。外傷誘發的水疱、疤痕與粟粒疹通常出現於肢端皮膚。
僅侵犯指甲之顯性失養型 EB (Dominant dystrophic EB nails only)
此疾病常只有在某位家庭成員因外傷誘發的皮膚水疱而就診、且回顧家系圖譜 (pedigree) 發現有一代或多代其他個體有指甲失養但無水疱、疤痕、粟粒疹或黏膜糜爛病史時才首次被診斷出來。因此,在某些個體中除了指甲失養之外並無其他臨床病徵,而指甲失養有時可僅限於大腳趾甲,故在許多家族中,根本不會懷疑為 dystrophic EB 的某一亞型。因此,此 dominant dystrophic EB 變異型很可能遠比目前所認知的更為常見。
脛前型顯性或隱性失養型 EB (Dominant or recessive dystrophic EB pretibial)
此 dystrophic EB 亞型可經由體染色體顯性(多數)或體染色體隱性傳遞而遺傳,外觀上有相當大的重疊。臨床上,於小腿前面有水疱、萎縮與疤痕(圖 4.20)。病灶通常呈紫紅色 (violaceous),有時類似扁平苔癬 (lichen planus)。手與足的指甲都傾向呈失養性。小腿可能會癢,其表型可與失養型 EB 癢疹型 (dystrophic EB pruriginosa)(見下節)重疊。臨床病徵並非僅限於脛部,在其他身體部位,尤其是骨性突起之上,可偵測到相對較輕微的皮膚脆弱、疤痕與粟粒疹。
癢疹型顯性或隱性失養型 EB (Dominant or recessive dystrophic EB pruriginosa)
此 dystrophic EB 亞型在臨床上與脛前型 (pretibial) 變異型重疊。主要差異在於劇烈的搔癢 (intense pruritus),其病因仍不確定。研究已排除合併的異位性體質 (atopy),以及在疾病致病機轉中一系列可能的代謝、生化與內分泌因素。其臨床特徵可類似肥厚性扁平苔癬 (hypertrophic lichen planus)、結節性癢疹 (nodular prurigo) 或自體免疫發炎性水疱病 (autoimmune inflammatory blistering),甚至人為性皮膚炎 (dermatitis artefacta)。如同脛前型亞型,症狀與病徵的初始發作可能延遲數十年,常導致皮膚病灶的遺傳病因被錯誤地排除。雖然脛部常受侵犯,搔癢性皮膚病灶可發生於任何部位。
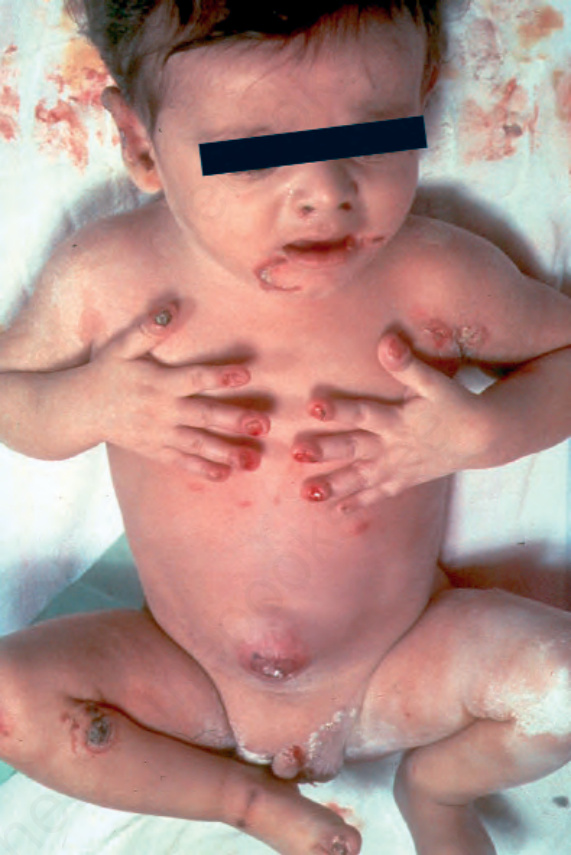
圖 4-14:泛發重度接合處型 EB (generalized severe junctional EB):剛出生的嬰兒有水疱與指甲侵犯。By courtesy of J. McGrath, MD, Institute of Dermatology, London, UK.
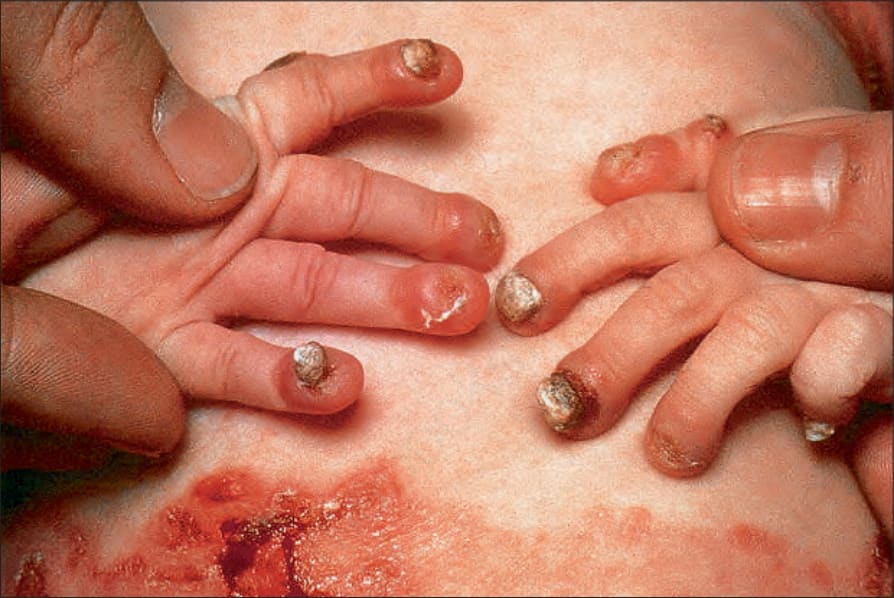
圖 4-15:泛發重度接合處型 EB (generalized severe junctional EB):嬰兒於癒合中水疱邊緣顯示肉芽組織 (granulation tissue)。By courtesy of the Institute of Dermatology, London, UK.
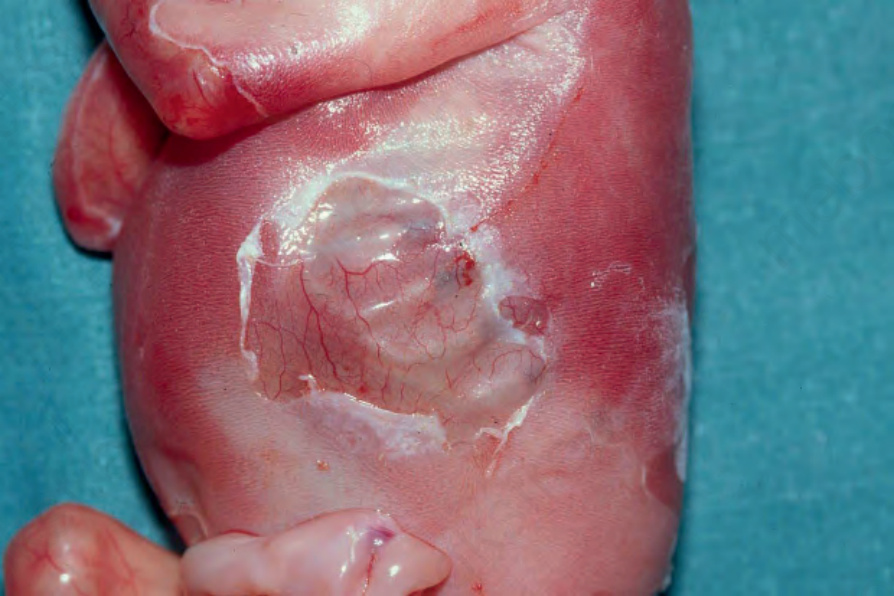
圖 4-16:合併幽門閉鎖之接合處型 EB (junctional EB with pyloric atresia):廣泛的水疱形成伴深部潰瘍。By courtesy of M.J. Tidman, MD, Institute of Dermatology, London, UK.
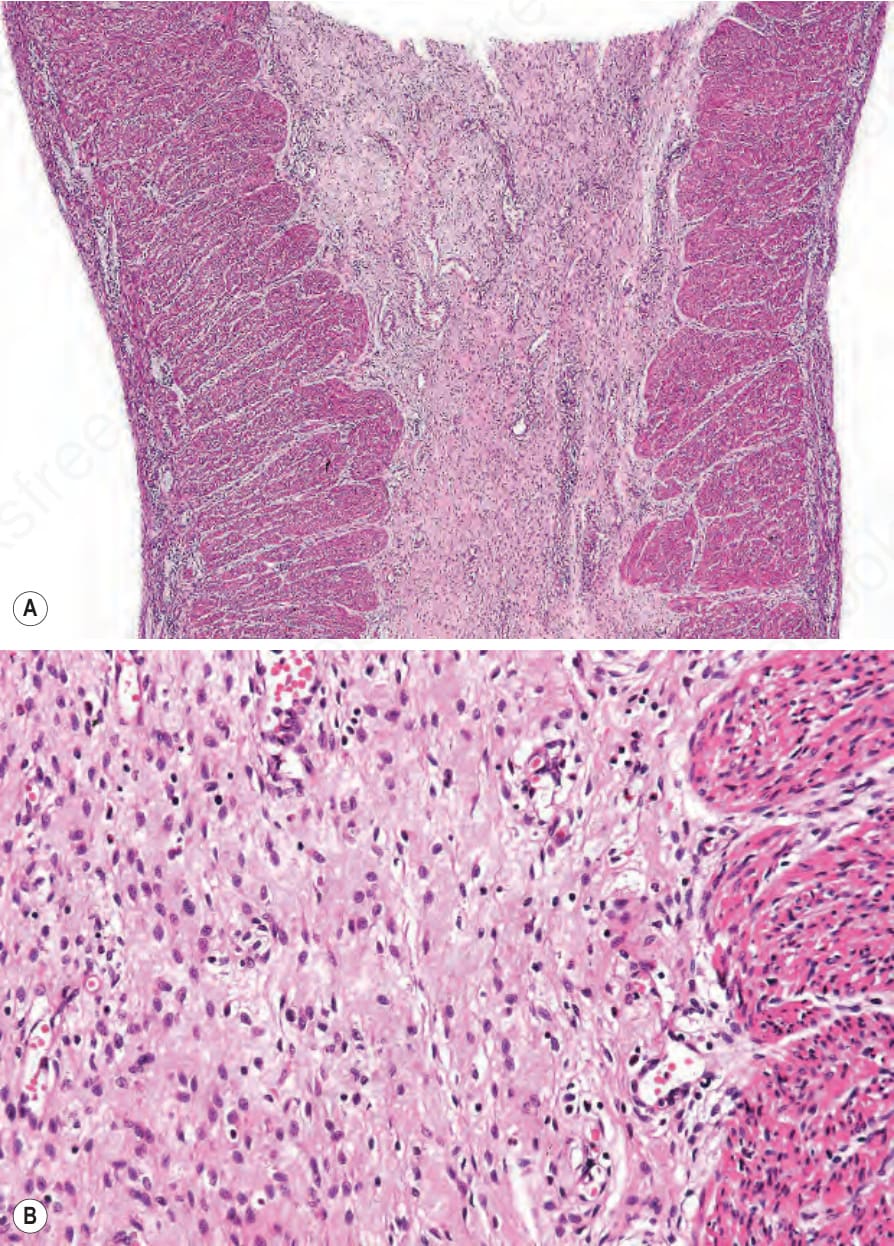
圖 4-17:(A, B) 合併幽門閉鎖之接合處型 EB (junctional EB with pyloric atresia):幽門管 (pyloric canal) 被纖維性結締組織 (fibrous connective tissue) 閉塞。
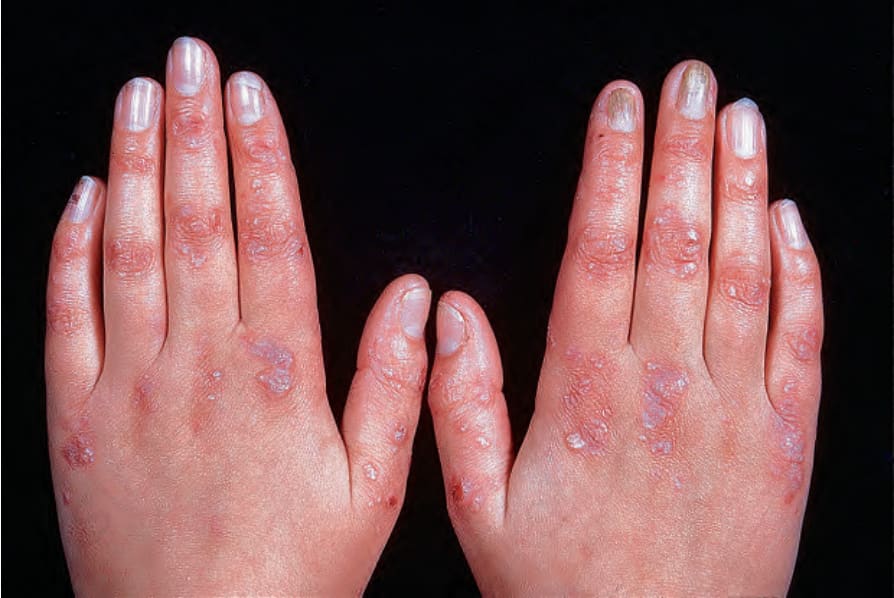
圖 4-18:泛發顯性失養型 EB (generalized dominant dystrophic EB):疤痕、粟粒疹 (milia) 與指甲失養 (nail dystrophy)。By courtesy of St John’s Institute of Dermatology, London, UK.
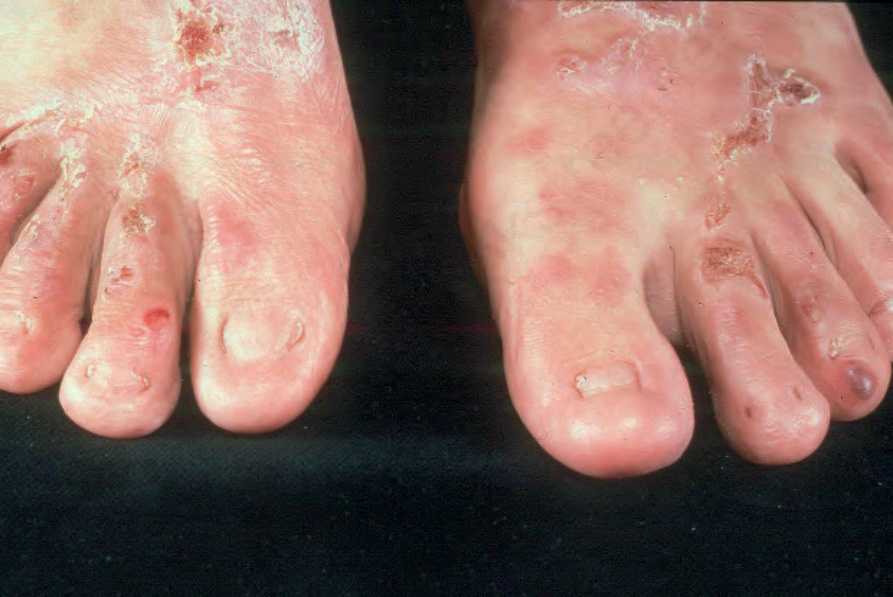
圖 4-19:肢端型顯性失養型 EB (dominant dystrophic EB–acral):以肢端為主的水疱與疤痕以及指甲失養。By courtesy of St John’s Institute of Dermatology, London, UK.
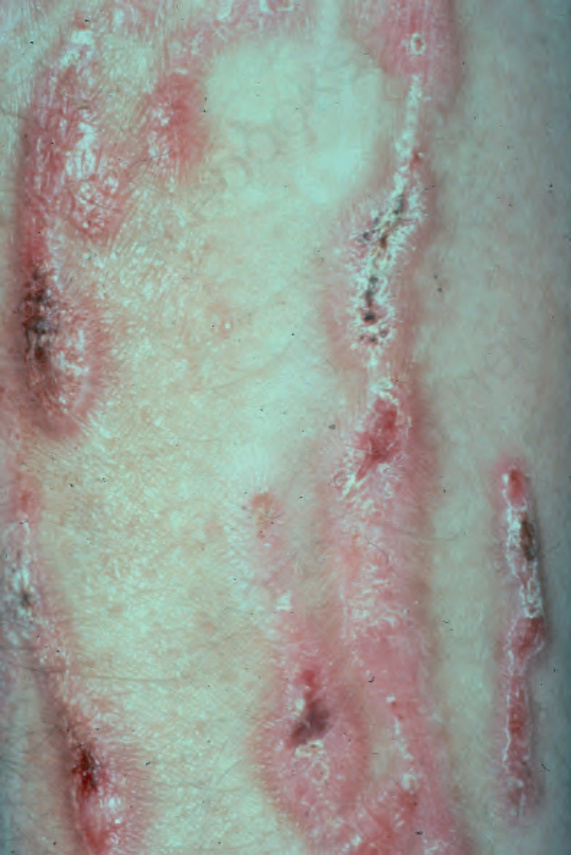
圖 4-20:脛前型顯性失養型 EB (dominant dystrophic EB–pretibial):局限於雙側脛前的線狀糜爛伴疤痕。By courtesy of St John’s Institute of Dermatology, London, UK.
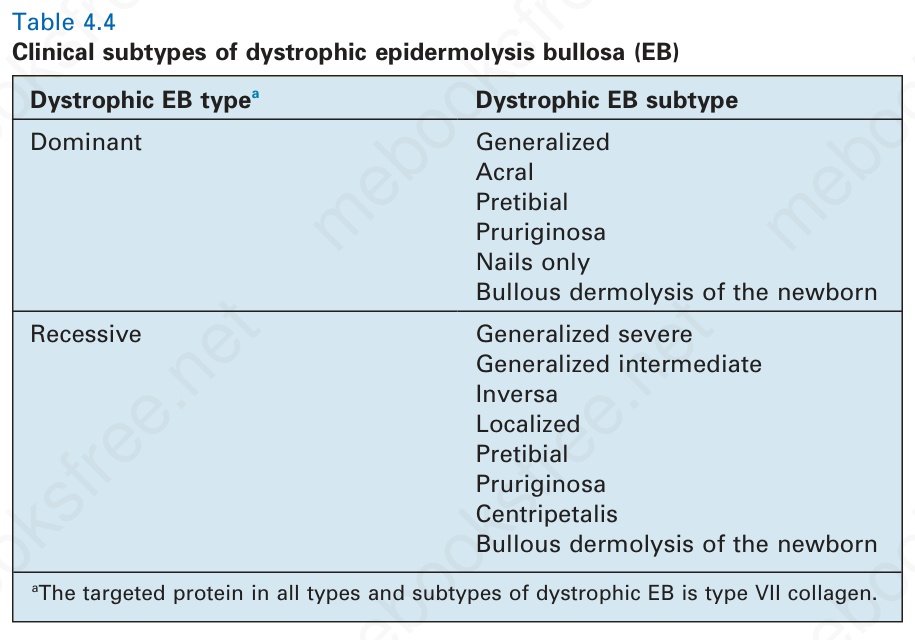
表 4-4:失養型表皮鬆解水疱症 (dystrophic epidermolysis bullosa, EB) 之臨床亞型。
失養型表皮鬆解水疱症續論 (Dystrophic EB, continued)
新生兒顯性或隱性失養型大疱病 (Dominant or recessive dystrophic bullous disease of the newborn)
dystrophic EB 一個奇特的變異型是:新生兒的水疱在出生後頭幾週或數月內顯示出自發性臨床改善的徵象(圖 4.21)。表型的改善與底層皮膚病理的改善相對應,在真皮-表皮交界處 (dermal–epidermal junction) 第 VII 型膠原蛋白 (type VII collagen) 增加。最初的皮膚切片顯示第 VII 型膠原蛋白於表皮內呈點狀標記 (punctate intraepidermal labeling),以及超微結構上呈現具顆粒外觀的擴張核周空泡 (dilated perinuclear vacuoles,星狀體 stellate bodies)。起初認為第 VII 型膠原蛋白的分泌與組裝為錨定纖維 (anchoring fibrils) 已完全矯正,因而有「新生兒暫時性大疱性真皮鬆解 (transient bullous dermolysis of the newborn)」的診斷標籤。然而,現今已認知到多數病例並未完全消退,且可能留有顯性或隱性 dystrophic EB 的永久性殘留病徵,儘管較生命早期為輕。
泛發重度隱性失養型 EB (Recessive dystrophic EB generalized severe)
多發性水疱於出生時即存在或出現於嬰兒期早期,且皮膚非常脆弱。臨床表現可能包括局部皮膚缺失,尤其影響小腿下段。水疱可在皮膚任何部位自發出現,或在最輕微的外傷後出現,且可能為出血性 (hemorrhagic)。粟粒疹 (milia) 與疤痕非常常見。雖然任何部位都可起水疱,主要區域為那些反覆遭受摩擦與其他形式物理外傷者。這些區域包括膝、肘、手、足、頸後、肩部與脊椎之上。慢性糜爛與潰瘍傾向被腐肉 (slough) 覆蓋,常伴有堆積的結痂與脫屑,增加續發性感染與生物膜 (biofilm) 形成的風險。搔癢與疼痛常見。頭皮常受侵犯,可能會發生疤痕性禿髮 (scarring alopecia)。在兒童期,反覆水疱形成伴進行性疤痕導致相鄰手指與腳趾的融合(「假性併指 pseudosyndactyly」)(圖 4.22)。指(趾)可發生進行性攣縮,並逐漸被一層繭狀 (cocoon-like) 的薄疤痕組織包覆,狀似連指手套 (mitten)。
口腔水疱與疤痕可導致舌繫帶過短 (ankyloglossia) 與小口畸形 (microstomia)。牙齦脆弱,輕柔刷牙即可能誘發上皮破壞並出血。舌乳頭 (lingual papillae) 消失,舌面變得平滑。雖然支持 dystrophic EB 存在原發性牙齒琺瑯質異常的證據有限,但牙齒發生齲齒 (caries) 的風險很高。食道水疱可能造成急性疼痛與吞嚥困難 (dysphagia),難以吞嚥固體食物,隨後因疤痕與纖維化造成的狹窄 (strictures) 而發展出阻塞。肛周水疱、糜爛與疼痛性裂隙 (fissures) 在兒童期常見。眼部併發症包括瞼球粘連 (symblepharon)、角膜糜爛,以及角膜混濁或疤痕。病人常有貧血 (anemic),骨質減少 (osteopaenia) 亦不少見。罕見情況下,續發性類澱粉沉積症 (secondary amyloidosis) 亦可發生於合併持續慢性發炎與廣泛疤痕的病例。此型 EB 一個常見且臨床上非常重要的併發症為鱗狀細胞癌 (squamous cell carcinoma) 的發生,甚至在年僅 6 歲的個體即可發生。多數癌症位於四肢,常在慢性、不癒合潰瘍的區域(圖 4.23)。多發性原發腫瘤、且每一次後續癌症的分化逐步喪失為其常見病程,死亡通常發生於第一個惡性腫瘤後 5 年內。
泛發中度隱性失養型 EB (Recessive dystrophic EB generalized intermediate)
有些較輕度的 dystrophic EB 亞型與 generalized severe recessive dystrophic EB 共有數項皮膚與皮膚外特徵,但程度輕微許多。皮膚與黏膜非常脆弱,但病灶(包括指甲變化、粟粒疹與萎縮性疤痕)傾向較為局限,且與許多 dominant dystrophic EB 病例所見者相似(圖 4.24)。生長遲滯與貧血通常輕微。假性併指 (pseudosyndactyly)、食道侵犯與鱗狀細胞癌亦可能發生,但這些併發症通常較輕微、較不頻繁,或與泛發性疾病相比發作較晚。
倒置型隱性失養型 EB (Recessive dystrophic EB inversa)
在倒置 (inversa) 亞型中,原發的水疱與疤痕區域包括腹股溝、腋窩、頸部與腰部區域,雖然在生命早期水疱的分布可能是泛發性的,而不能預示其後續型態。外傷性角膜糜爛與食道病灶常見。指甲失養、黏膜侵犯與牙齒變化與本疾病泛發型者相似。病人亦有發展出鱗狀細胞癌的風險。屈側侵犯居多的原因尚不清楚。
局限型隱性失養型 EB (Recessive dystrophic EB localized)
recessive dystrophic EB 的局限型與 generalized intermediate 亞型重疊,亦與歸類為倒置型 (inversa)、脛前型 (pretibial) 或僅侵犯指甲型 (nails only) 者重疊,凸顯了臨床特徵的廣泛光譜,以及試圖將所有亞型塞進整齊、明確界定之類別這一目標的某種武斷性。皮膚脆弱、疤痕與粟粒疹大多侷限於手、足與指甲,且可能為次要特徵。在某些病例中,症狀的發作可能延遲至出生後數年。黏膜侵犯罕見。
向心型隱性失養型 EB (Recessive dystrophic EB centripetalis)
此 dystrophic EB 亞型罕見,且因僅有一名個體被報導過,未來分類中很可能會被併入其他類別。該被報導的個體於出生時有泛發性水疱,但在生命第一年內其皮膚病活性即侷限於手與足。歷經數十年,水疱以向心 (centripetal) 的方式緩慢地向近端進展,活躍的水疱與粟粒疹僅沿活躍邊緣分布,而遠端則有萎縮性疤痕與指甲失養。未發生全身性侵犯。
Kindler 症候群 (Kindler syndrome)
Kindler 症候群 (Kindler syndrome) 為一種體染色體隱性疾病。起初它可類似泛發型或局限型的 dystrophic EB,雖然皮膚切片典型顯示可變的裂隙平面 (variable plane of cleavage) 與破碎或重複的緻密板 (fragmented or duplicated lamina densa),與 EB simplex、junctional EB 及 dystrophic EB 所根據的特定組織分離平面形成對比。因此,Kindler syndrome 被歸類為 EB 的一個獨立類別(表 4.1)。臨床上,最初的皮膚水疱在兒童期減輕,取而代之的是發展出進行性異色症 (progressive poikiloderma) 的病徵(圖 4.25)。在此階段,鑑別診斷包括其他先天性異色症性疾病,包括先天性角化不良 (dyskeratosis congenita) 與 Rothmund-Thomson syndrome。其他特徵包括牙齦發炎 (gingival inflammation)、瞼外翻 (ectropion)、角膜糜爛、慢性結腸炎 (chronic colitis)、牙周病 (periodontal disease)、外尿道口疤痕 (scarring of the external urethral meatus),以及發展出皮膚鱗狀細胞癌的風險增加。雙手亦可顯示假性併指 (pseudosyndactyly) 的證據,類似某些 dystrophic EB 病例。
EB 的病理基礎 (Pathologic Basis of EB)
在 EB 各亞型中涉及的兩個關鍵黏附複合體 (adhesion complexes) 為半橋粒 (hemidesmosome)(圖 4.26)與橋粒 (desmosome)(圖 4.27),而涉及不同型態 EB 的 18 個不同基因列於表 4.5。
角蛋白 5 與 14:KRT5、KRT14 (Keratins 5 and 14: KRT5, KRT14)
角蛋白 (keratins) 是上皮細胞細胞質中含量最豐富的結構蛋白。成對的角蛋白單體 (keratin monomers) 聚合形成直徑 10 nm 的中間絲 (intermediate filaments) 網絡,以維持角質形成細胞 (keratinocytes) 的形狀。Keratin 5 與 14 是基底層角質形成細胞中主要的角蛋白。KRT5 或 KRT14 的體染色體顯性突變是 EB 最常見亞型——局限型單純型 EB (localized EB simplex) 的基礎,此疾病全球影響近 400 000 人。EB simplex 通常導致輕微的水疱,典型在夏季月份較嚴重,且不會留下疤痕。然而,亦有數種其他 EB simplex 的臨床變異型同樣肇因於 KRT5 或 KRT14 突變(表 4.2);多數為顯性,但 KRT14 的體染色體隱性突變也可能發生。水疱典型發生於基底層角質形成細胞層內(圖 4.28),且某些 KRT5 或 KRT14 突變可能造成角蛋白絲網絡 (keratin filament network) 的結構改變(圖 4.29)。以石蠟包埋切片 (paraffin-embedded sections) 的光學顯微鏡診斷 EB simplex 可能具挑戰性,因為早期變化發生於非常接近真皮-表皮交界處之處且可能很細微(圖 4.30),而較舊的病灶則可能呈現為表皮下 (subepidermal)(圖 4.31)。
斑菲素蛋白-1:PKP1 (Plakophilin-1: PKP1)
某些型態的 EB 肇因於橋粒細胞-細胞接合 (desmosome cell–cell junctions) 內所見蛋白質的突變(圖 4.32)。橋粒 (desmosomes) 在相鄰細胞之間形成結構性與訊號傳遞性的連結,可見於皮膚角質形成細胞、心肌細胞 (cardiac myocytes)、腦膜 (meninges) 與淋巴結皮質 (cortex of lymph nodes)。Plakophilin-1 的定位侷限於角質形成細胞的橋粒,而 PKP1 的體染色體隱性功能喪失型 (loss-of-function) 突變導致外胚層發育不良-皮膚脆弱症候群 (ectodermal dysplasia – skin fragility syndrome)。水疱與糜爛肇因於角質形成細胞內橋粒內斑塊 (desmosomal inner plaque) 處角質形成細胞黏附的喪失(即非真正的棘層鬆解 acantholysis);外胚層發育不良部分肇因於表皮分化與增生的改變,但也因為 plakophilin-1 在其他組織中亦具有細胞核訊號傳遞 (nuclear signaling) 的角色。
斑珠蛋白:JUP (Plakoglobin: JUP)
(接前段)伴有泛上皮 (pan-epidermal) 棘層鬆解與不良預後。在合併 Naxos 病——羊毛狀髮 (woolly hair)、掌蹠角化症 (palmoplantar keratoderma) 與心肌病變 (cardiomyopathy) 之組合——的個體中亦有體染色體隱性突變的報導;異型合子帶因者 (heterozygous carriers) 亦可能易發生心律不整 (cardiac arrhythmias) 或心臟衰竭。其他體染色體顯性突變可能造成心肌病變。
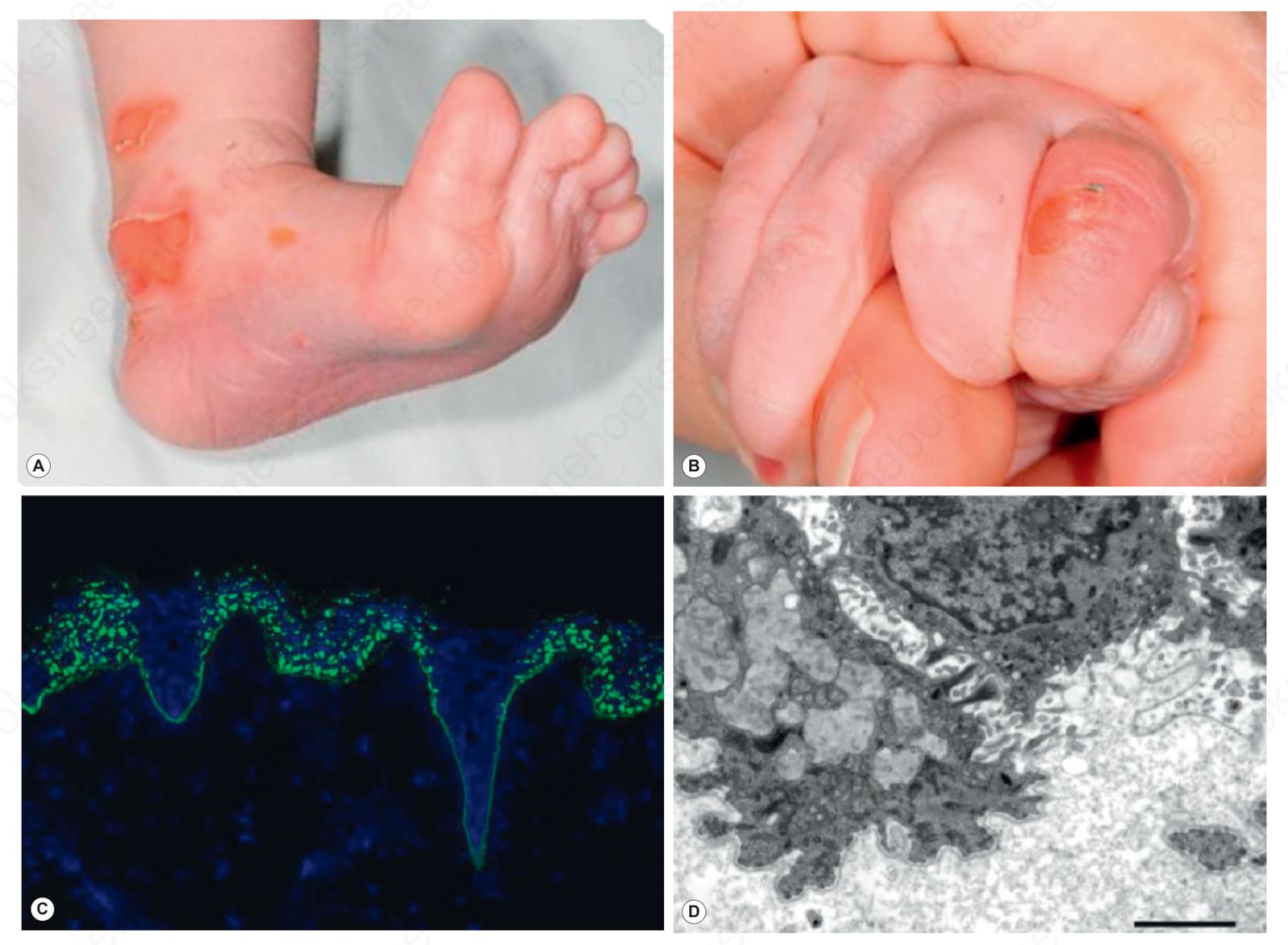
圖 4-21:新生兒大疱性真皮鬆解 (bullous dermolysis of the newborn):(A) 足跟的水疱;(B) 手指的水疱;(C) 第 VII 型膠原蛋白 (type VII collagen) 存在於真皮-表皮交界處 (dermal–epidermal junction),但表皮內亦有顯著的點狀染色 (punctate staining);(D) 超微結構上,此基底層角質形成細胞內有眾多淡灰色星狀體 (stellate bodies,含第 VII 型膠原蛋白的擴張高基氏體 dilated Golgi apparatus)。此型 dystrophic EB 通常傾向於在生命頭幾個月內自發改善。
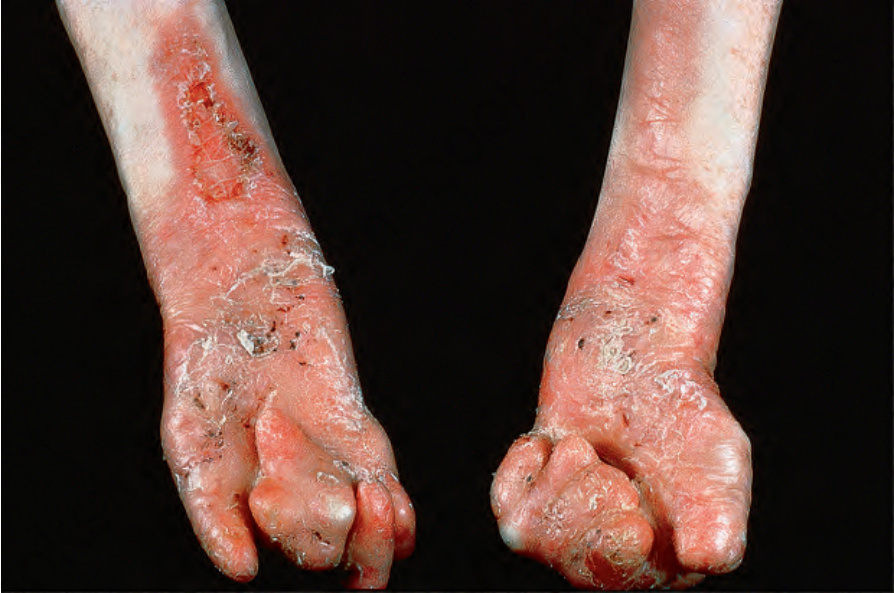
圖 4-22:泛發重度隱性失養型 EB (generalized severe recessive dystrophic EB):除了嚴重的連指手套畸形 (mitten deformity) 外,還有非常嚴重的疤痕與脫屑。By courtesy of St John’s Institute of Dermatology, London, UK.
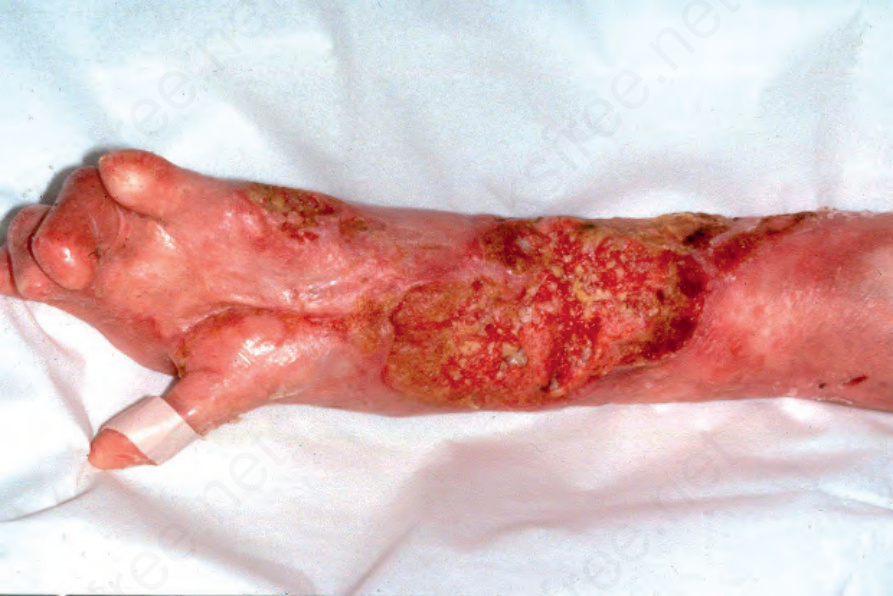
圖 4-23:泛發重度隱性失養型 EB (generalized severe recessive dystrophic EB):此病人可見眾多大型角化病 (keratoses)。其中許多會進展為鱗狀細胞癌 (squamous cell carcinoma)。Courtesy of R.A.J. Eady, MD, and B. Mayou, MD, St Thomas’ Hospital, London, UK.
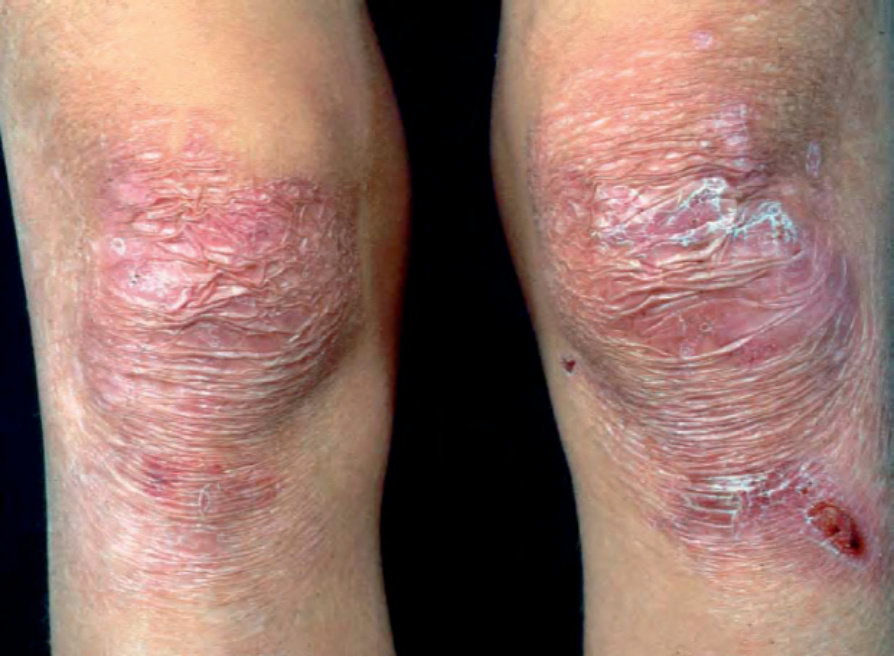
圖 4-24:泛發中度隱性失養型 EB (generalized intermediate recessive dystrophic EB):此個體於雙膝之上有萎縮性疤痕與近期的糜爛。
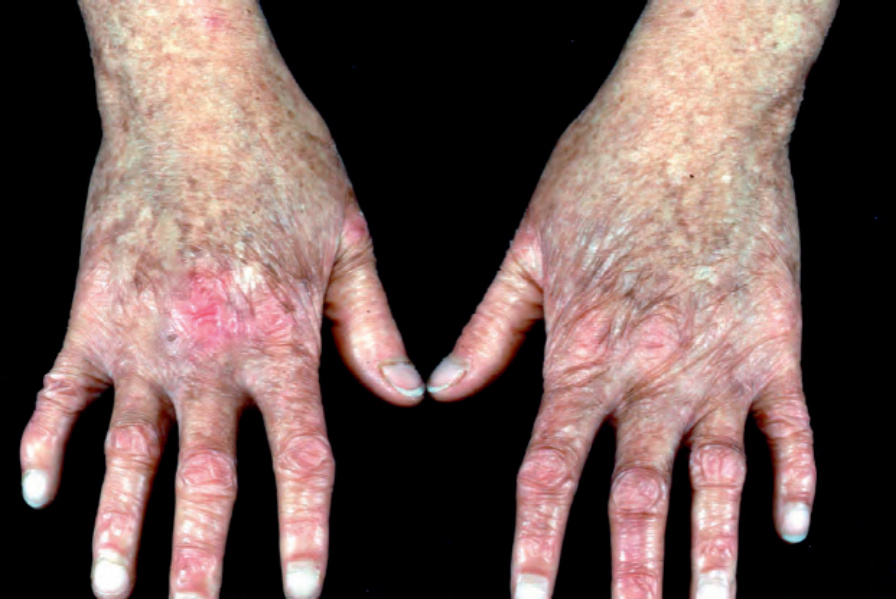
圖 4-25:Kindler 症候群 (Kindler syndrome):此 14 歲女孩的雙手顯示異色症 (poikiloderma,色素過度沉著、色素減退、萎縮與微血管擴張 telangiectasias)。
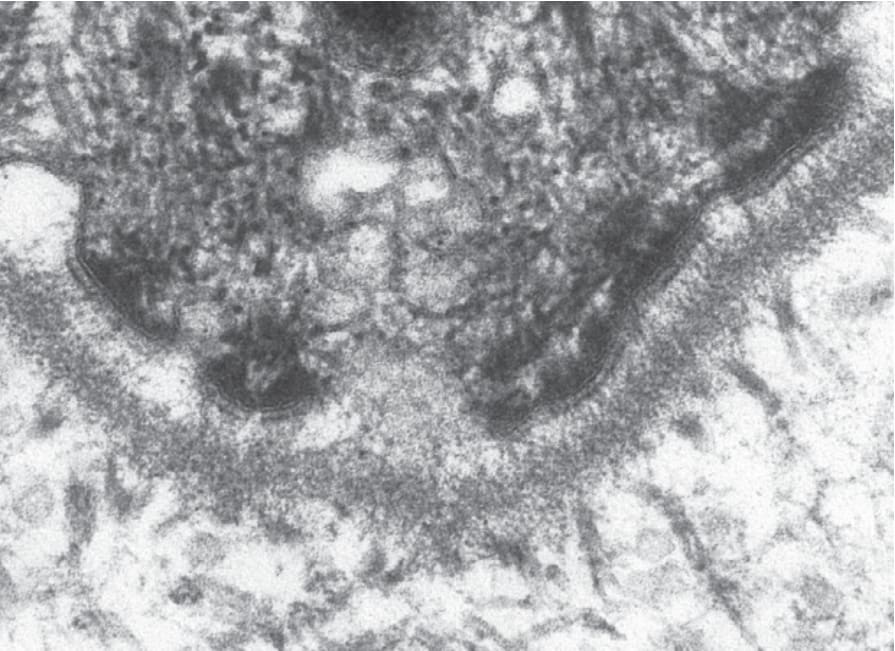
圖 4-26:正常人類皮膚中真皮-表皮交界處半橋粒 (hemidesmosome) 的超微結構外觀。
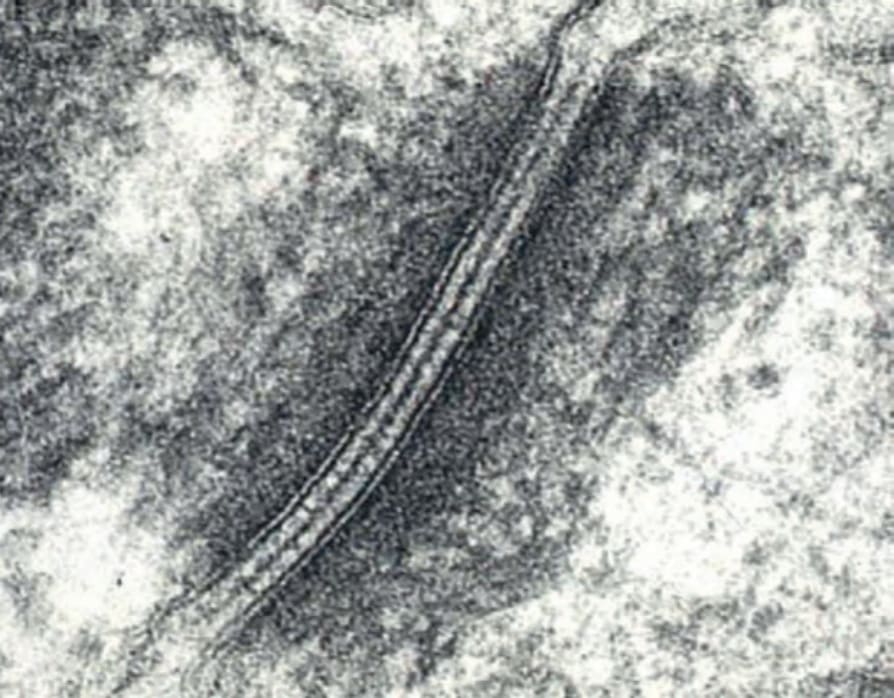
圖 4-27:正常人類皮膚中兩個角質形成細胞之間橋粒 (desmosome) 的超微結構外觀。
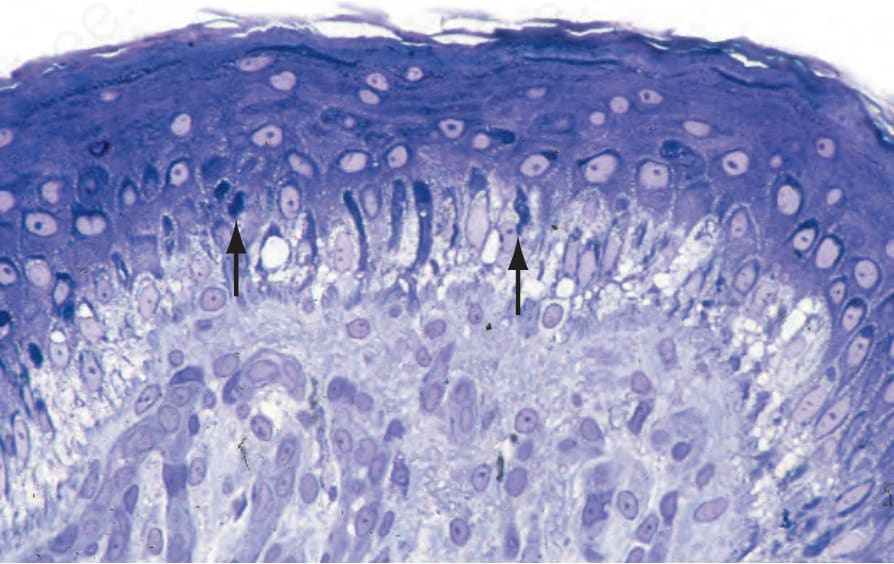
圖 4-28:泛發重度單純型 EB (generalized severe EB simplex):相鄰的臨床上正常皮膚中存在眾多張力絲團塊 (tonofilament clumps)(箭頭所示)。By courtesy of J.A. McGrath, MD, St John’s Institute of Dermatology, London, UK.
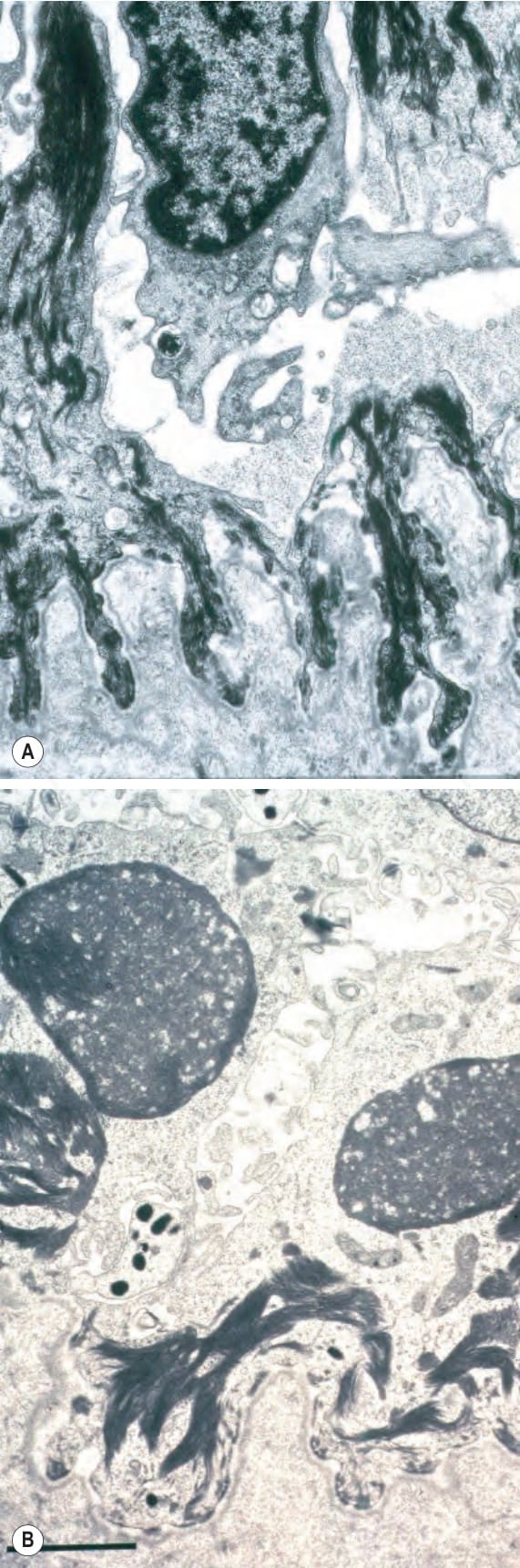
圖 4-29:泛發重度單純型 EB (generalized severe EB simplex):(A) 電子顯微鏡照片顯示角質形成細胞內裂開 (intrakeratinocyte splitting);(B) 張力絲團塊 (tonofilament clumps) 的近距離視野。By courtesy of J.A. McGrath, MD, and R.A.J. Eady, MD, St John’s Institute of Dermatology, London, UK.
圖 4-30:單純型 EB (EB simplex):水疱發展中最早的組織學特徵為基底層角質形成細胞的明顯空泡化 (vacuolation),即所謂的細胞溶解 (cytolysis)。
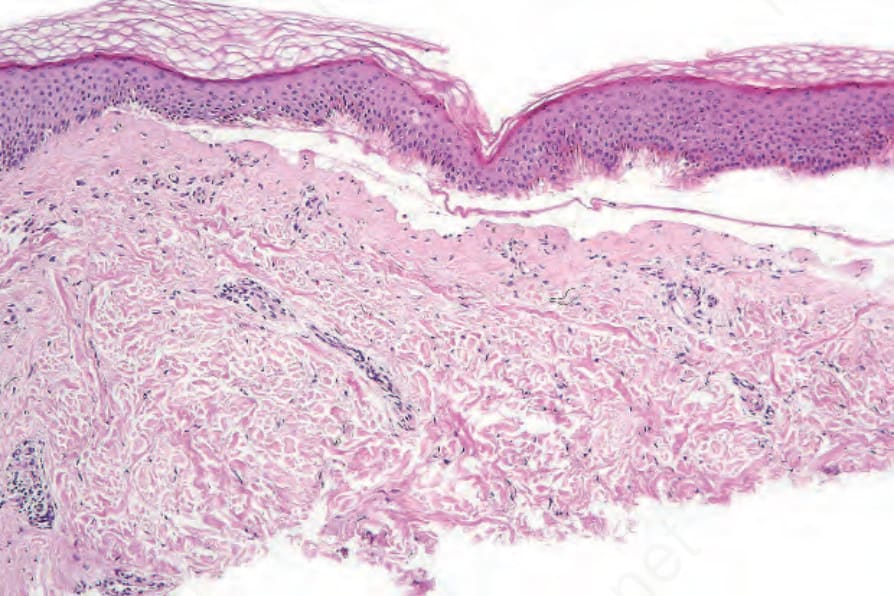
圖 4-31:單純型 EB (EB simplex):舊病灶;其特徵為無細胞的表皮下水疱 (cell-free subepidermal blister),並無特異性。
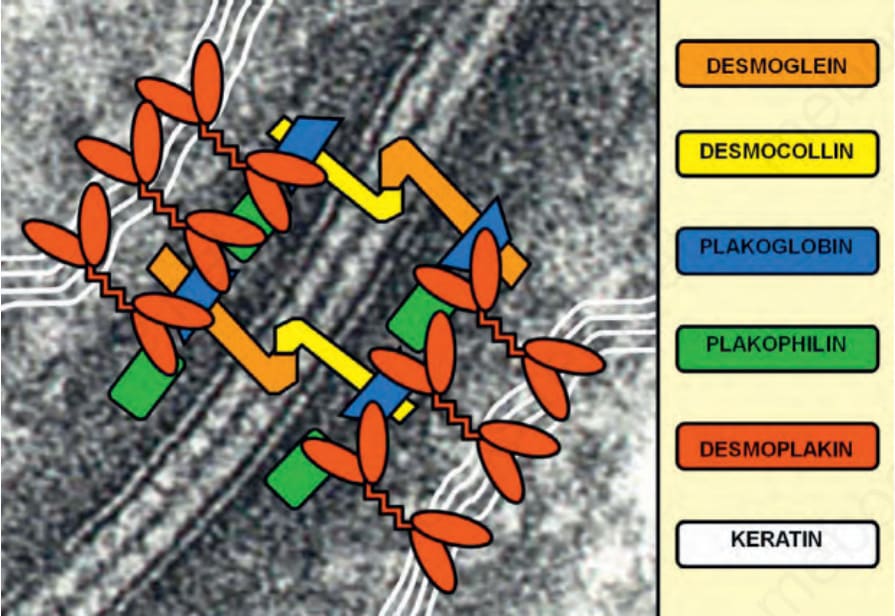
圖 4-32:橋粒 (desmosomes) 跨膜與細胞內組成成分的示意圖,其在相鄰角質形成細胞的角蛋白絲網絡 (keratin filament networks) 之間提供橋接。
EB 的分子病理續論 (Molecular Basis of EB, continued)
親外泌蛋白-5:EXPH5 (Exophilin-5: EXPH5)
EXPH5 編碼親外泌蛋白-5 (exophilin-5,亦稱為 Slac2-b),為 Rab GTPase Rab27B 的一個效應蛋白 (effector protein),被認為在沿著肌動蛋白 (actin) 與微管蛋白 (tubulin) 網絡的細胞內囊泡運輸 (intracellular vesicle trafficking),以及囊泡向細胞膜的轉運中具有重要角色。EXPH5 的功能喪失型 (loss-of-function) 突變導致角蛋白絲團塊化 (keratin filament clumping)、細胞溶解 (cytolysis)、棘層鬆解 (acantholysis) 與細胞質(核周)囊泡 (cytoplasmic perinuclear vesicles) 增加。此型遺傳性皮膚脆弱已被歸類為一種體染色體隱性型的 EB simplex。
轉麩醯胺酸酶 5:TGM5 (Transglutaminase 5: TGM5)
TGM5 的體染色體隱性突變是肢端脫皮症候群 (acral peeling skin syndrome) 的基礎。Transglutaminase 5 是皮膚中所表現的八種不同轉麩醯胺酸酶 (transglutaminase) 酵素之一,在角質化細胞膜套 (cornified cell envelope) 的形成中具有獨特的角色。水疱形成的層面發生於顆粒層 (granular layer) 之上、恰位於角質層 (stratum corneum) 下方。然而,鑑於手掌與足底的角質層相對於其他身體部位較厚,其臨床外觀常類似最常見型態的 localized EB simplex,且確實可能造成某些臨床混淆,雖然兩者皆被納入最新的 EB 分類中(表 4.2)。
網蛋白:PLEC (Plectin: PLEC)
Plectin 是一種表皮斑蛋白 (plakin) 蛋白,亦見於橫紋肌的 z 線 (z-lines) 內。PLEC 的體染色體隱性突變造成合併肌肉失養症的 EB simplex (EB simplex associated with muscular dystrophy),表現為相對輕微的皮膚水疱與進行性肌肉無力。PLEC 的體染色體隱性突變亦可造成合併幽門閉鎖的皮膚水疱 (skin blistering with pyloric atresia),或偶爾兩種表現皆有,或有時僅有皮膚水疱。Plectin 的體染色體顯性突變亦可能發生於其他型態的 localized EB simplex(佔所有病例最多 10%),包括目前 EB 分類中少數以人名命名的變異型之一——Ogna 亞型。
橋粒斑蛋白:DSP (Desmoplakin: DSP)
Desmoplakin 是橋粒斑塊 (desmosomal plaque) 主要的細胞內組成成分。體染色體隱性突變可導致毀滅性的黏膜皮膚脆弱 (mucocutaneous skin fragility),尤其是一種嚴重的棘層鬆解型 EB (severe acantholytic form of EB)。官方上此疾病被歸類為一種基底上型 (suprabasal) 的 EB simplex,雖然 desmoplakin 具有泛表皮 (pan-epidermal) 的表現,且表皮所有層次皆顯示棘層鬆解(圖 4.33),超微結構上裂隙穿過橋粒的內斑塊 (inner plaque of desmosomes)(圖 4.34)。在這些病例中 desmoplakin 表現的喪失導致早期死亡,因為深層的皮膚喪失與其他器官(尤其是心臟)的潛在侵犯。其他體染色體隱性突變可能導致羊毛狀髮 (woolly hair) 與角化症 (keratoderma) 但無皮膚脆弱,而 DSP 的體染色體顯性突變亦可引起條紋狀掌蹠角化症 (striate palmoplantar keratoderma) 或致心律不整性心肌病變 (arrhythmogenic cardiomyopathy)。
肌張力蛋白表皮異構物 (BP230):DST-e (Dystonin epidermal isoform (BP230): DST-e)
肌張力蛋白 (dystonin) 表皮異構物(亦稱為 230 kDa 大疱性類天疱瘡抗原 BP230)的體染色體隱性突變,導致一種相對輕度的 EB simplex。超微結構上,半橋粒內斑塊 (hemidesmosomal inner plaques)——即角蛋白中間絲錨定至半橋粒之處——完全缺失。雖然 dystonin 異構物具有廣泛的組織分布,神經學或心臟侵犯似乎並非臨床特徵——以肢端為主的皮膚水疱才是主要的異常。
α6β4 整合素:ITGA6、ITGB4 (α6β4 integrin: ITGA6, ITGB4)
α6β4 整合素 (α6β4 integrin) 是一種參與半橋粒組裝以及上皮-間質訊號傳遞 (epithelial–mesenchymal signaling) 的細胞黏附二聚體 (dimer)。ITGA6 或 ITGB4(分別編碼 α6 與 β4 整合素次單元)的突變導致合併幽門閉鎖的體染色體隱性 junctional EB。皮膚脆弱與胃部阻塞程度兩者的臨床嚴重度皆可不同,但通常需要手術矯正幽門。較嚴重型態的疾病肇因於 ITGA6 或 ITGB4 兩個對偶基因 (alleles) 皆有功能喪失型突變,雖然某些關鍵半胱胺酸殘基 (cysteine residues) 的錯義突變 (missense mutations) 亦可能造成毀滅性的臨床後果。其他錯義突變可導致不同型態的 generalized intermediate junctional EB。
斑珠蛋白:JUP (Plakoglobin: JUP)
Plakoglobin 是橋粒的一種細胞內犰狳蛋白 (armadillo protein) 組成成分。體染色體隱性突變可造成一種棘層鬆解型的 EB simplex。這些病例與某些 DSP 突變相似——此疾病被歸類為基底上型 (suprabasal) 的 EB simplex,但伴有泛表皮 (pan-epidermal) 的棘層鬆解與不良預後。在合併 Naxos 病——羊毛狀髮 (woolly hair)、掌蹠角化症 (palmoplantar keratoderma) 與心肌病變 (cardiomyopathy) 之組合——的個體中亦有體染色體隱性突變的報導;異型合子帶因者 (heterozygous carriers) 亦可能易發生心律不整或心臟衰竭。其他體染色體顯性突變可能造成心肌病變。
α3 整合素次單元:ITGA3 (α3 integrin subunit: ITGA3)
α3 整合素次單元 (α3 integrin subunit) 是真皮-表皮交界處局部接觸 (focal contacts) 的一個組成成分,於此處它可與 β1 integrin 形成二聚體,並促成上皮-間質訊號傳遞。ITGA3 的體染色體隱性功能喪失型突變已於合併肺部發炎 (pulmonary inflammation) 與先天性腎病症候群 (congenital nephrotic syndrome) 的個體中被報導,反映了 α3 integrin 在肺與腎生物學中的重要角色。值得注意的是,皮膚水疱相對輕微且不一定存在。因為肺/腎疾病,預後不佳。
賴丁蛋白-1:KIND1/FERMT1 (Kindlin-1: KIND1/FERMT1)
賴丁蛋白-1 (Kindlin-1,亦稱為 fermitin family homolog-1) 是真皮-表皮交界處局部接觸 (focal contacts) 的一個組成成分,在肌動蛋白細胞骨架 (actin cytoskeleton) 的錨定,以及經由 β1 integrin 形成訊號傳遞平台中具有角色。KIND1/FERMT1 的體染色體隱性突變導致 Kindler 症候群 (Kindler syndrome),這是一種水疱性遺傳性皮膚病 (blistering genodermatosis),在生命早期可能類似 dystrophic EB,但隨年齡增長,水疱常減少,並發展出新的特徵——光敏感 (photosensitivity) 與異色症 (poikiloderma,色素過度沉著、色素減退、微血管擴張 telangiectases 與皮膚萎縮之組合),主要在日曬部位。患有 Kindler syndrome 的個體亦有鱗狀細胞癌風險增加。
第 XVII 型膠原蛋白:COL17A1 (Type XVII collagen: COL17A1)
第 XVII 型膠原蛋白 (type XVII collagen,亦稱為 180 kDa 大疱性類天疱瘡抗原),是一種位於半橋粒與透明板 (lamina lucida) 內的跨膜蛋白。它是自體免疫水疱病大疱性類天疱瘡 (bullous pemphigoid) 中的抗原標的,但兩個對偶基因皆有功能喪失型突變則導致 generalized intermediate junctional EB(先前稱為 non-Herlitz 或 generalized atrophic benign EB)。某些第 XVII 型膠原蛋白的顯性錯義突變可能導致牙齒琺瑯質缺損與偶爾的皮膚脆弱,但 COL17A1 中多數致病性突變為體染色體隱性。
層黏連蛋白-332:LAMA3、LAMB3、LAMC2 (Laminin-332: LAMA3, LAMB3, LAMC2)
層黏連蛋白-332 (Laminin-332,先前稱為 laminin-5) 是一種異三聚體蛋白 (heterotrimeric protein),由 α3、β3 與 γ2 層黏連蛋白多肽鏈組成,位於表皮基底膜 (epidermal basement membrane) 的透明板/緻密板 (lamina lucida/lamina densa) 內。體染色體隱性突變可引起 generalized severe(先前稱為 Herlitz junctional EB)、generalized intermediate,或更局限型態的 junctional EB(表 4.3)。組織裂隙的平面穿過真皮-表皮交界處的透明板(圖 4.35)。Generalized severe 疾病與廣泛的黏膜皮膚脆弱及不良預後相關。臨床上較不嚴重型態的 junctional EB 通常與允許殘留某些功能性 laminin-332 蛋白的突變相關。LAMA3 基因 LAMA3A 異構物的突變與喉-甲-皮症候群 (laryngo-onycho-cutaneous syndrome) 相關,其過度的肉芽組織可導致喉部阻塞與失明。
第 VII 型膠原蛋白:COL7A1 (Type VII collagen: COL7A1)
第 VII 型膠原蛋白 (type VII collagen) 是錨定纖維 (anchoring fibrils) 的主要組成成分,錨定纖維是插入緻密板 (lamina densa) 真皮側的黏附複合體,並有真皮膠原纖維穿越其間,以確保表皮與真皮之間的黏附(圖 4.36)。COL7A1 的突變是 dystrophic EB 體染色體顯性與隱性型態兩者的基礎。典型而言,COL7A1 兩個對偶基因皆有功能喪失型突變是 generalized severe recessive dystrophic EB 的基礎,其錨定纖維在結構上有缺陷或完全缺失(圖 4.37)。不良的傷口癒合導致慢性傷口、毀容性疤痕形成 (mutilating scar formation),以及鱗狀細胞癌發生率增加,常在數年期間出現多發性原發腫瘤(圖 4.38 與 4.39)。初始腫瘤可能分化良好或為疣狀 (verrucous) 且難以與假性上皮瘤樣增生 (pseudoepitheliomatous hyperplasia) 區分,雖然後續的腫瘤會逐漸變得分化較差。然而,存在著一個臨床嚴重度光譜,破壞性較低的突變會引起較不嚴重的中度或局限型表型(表 4.4)。Dominant dystrophic EB 在臨床上通常比隱性疾病輕微,且多數病例肇因於第 VII 型膠原蛋白三股螺旋 (triple helix) 內的異型合子錯義突變 (heterozygous missense mutations)。
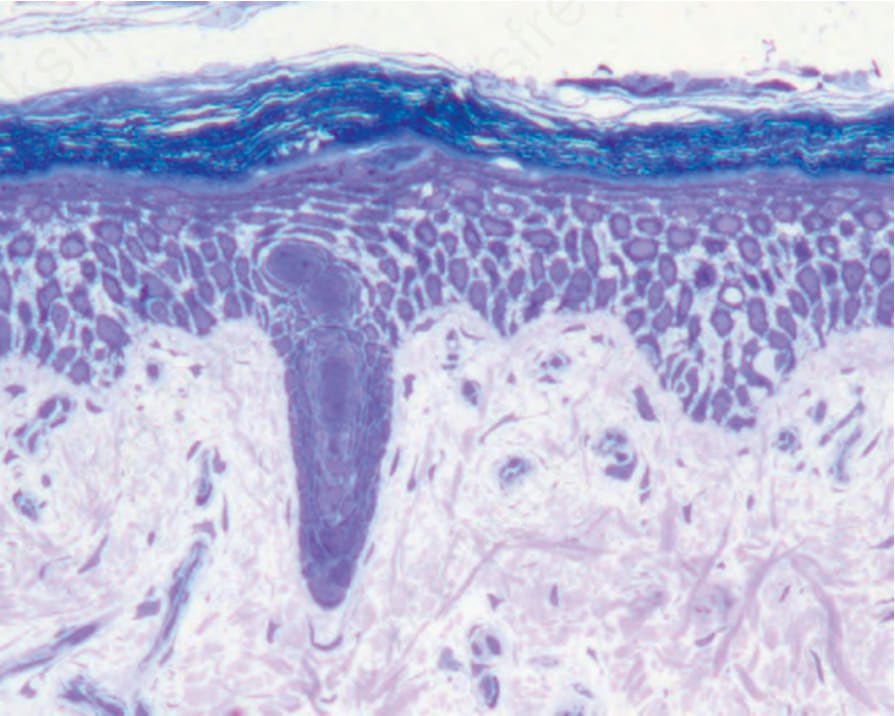
圖 4-33:橋粒型單純型 EB (desmosomal EB simplex):表皮內泛表皮的細胞-細胞脫離 (pan-epidermal cell-cell detachment),此處肇因於 desmoplakin 的體染色體隱性突變。
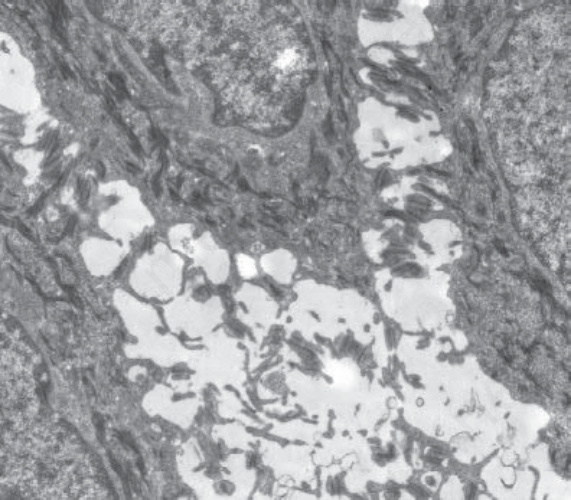
圖 4-34:橋粒型單純型 EB (desmosomal EB simplex):導致細胞分離的超微結構裂隙平面穿過細胞內橋粒斑塊 (intracellular desmosomal plaque),與此病例中突變 desmoplakin 的定位一致。
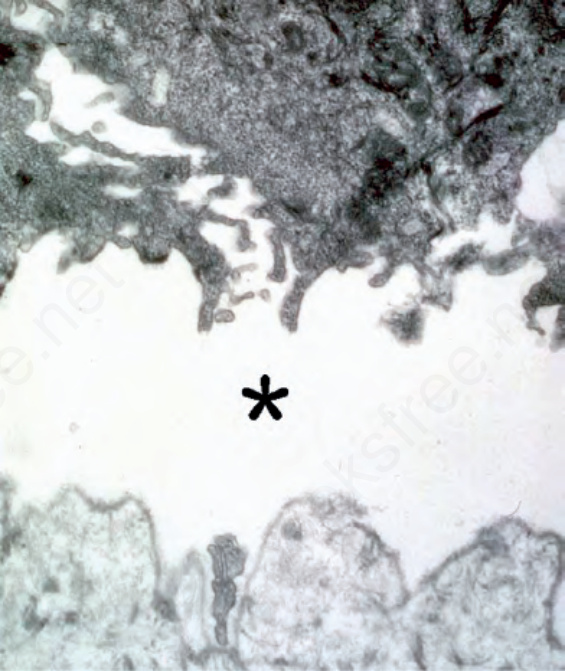
圖 4-35:接合處型 EB (junctional EB):真皮-表皮交界處水疱形成的層面穿過透明板 (lamina lucida)(星號處)。
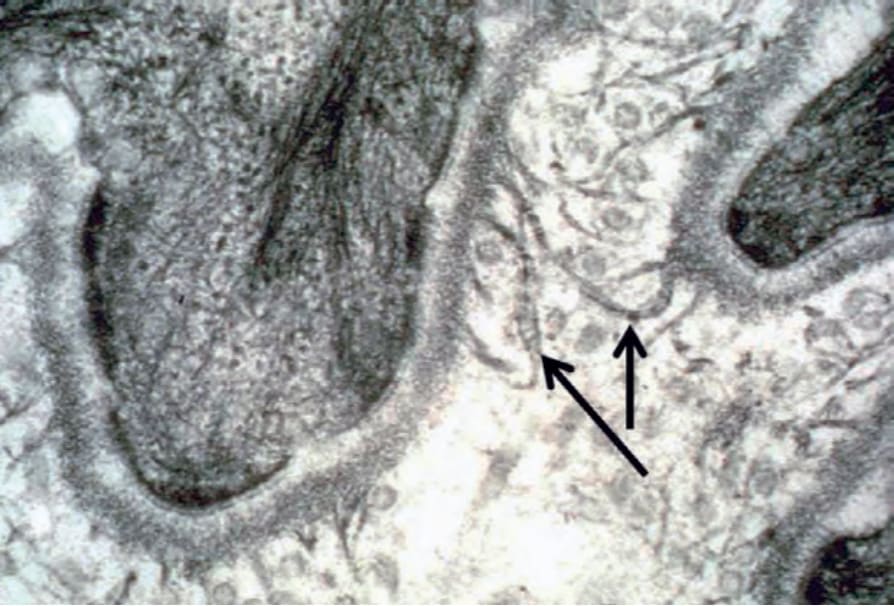
圖 4-36:正常皮膚中的錨定纖維 (anchoring fibrils):具扇形外觀、中央橫帶 (central cross-banding) 並插入緻密板 (lamina densa) 的纖維狀結構,代表淺層真皮中錨定纖維的超微結構標誌(箭頭所示)。
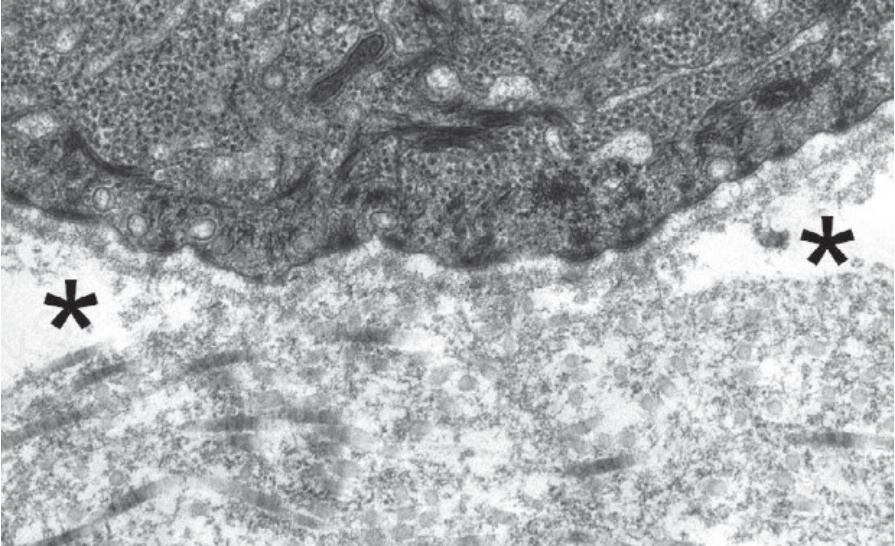
圖 4-37:泛發重度隱性失養型 EB (generalized severe recessive dystrophic EB):緻密板 (lamina densa) 下方完全缺乏錨定纖維,伴有緻密板下水疱形成 (sub-lamina densa blistering) 的發生(星號處)。
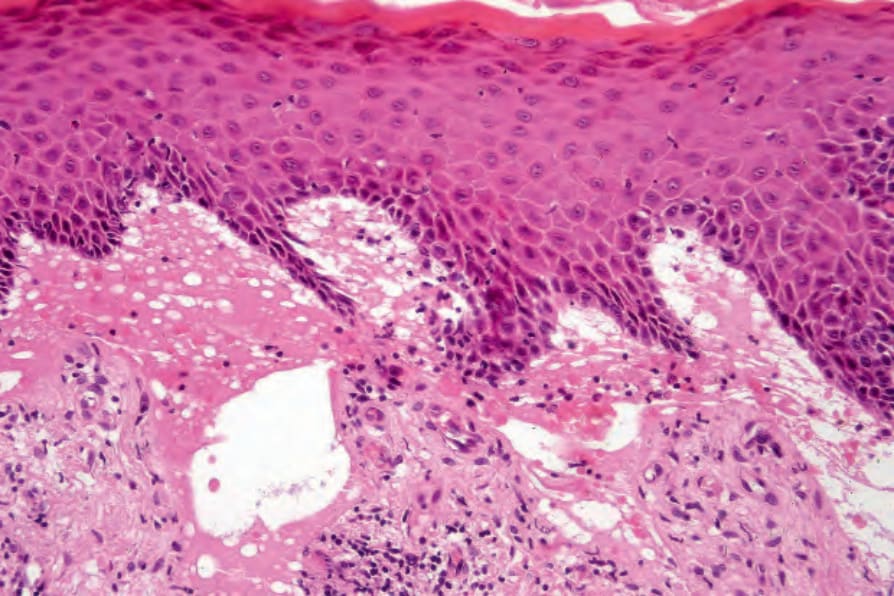
圖 4-38:泛發重度隱性失養型 EB (generalized severe recessive dystrophic EB):除了明顯的表皮下水疱形成 (subepidermal blistering) 之外,還有真皮疤痕與慢性發炎。