Epidermolysis bullosa
Epidermolysis bullosa
Epidermolysis bullosa (EB) refers to a heterogeneous group of diseases in which the skin and sometimes the mucous membranes blister easily in response to mild trauma, hence the alternative title ‘mechanobullous disease’, which has sometimes been applied.1 Indeed, the descriptive term
119 Epidermolysis bullosa
K5, K14 (IF)
300K>IFAR
BP230 Plectin
a6
HD
CM
b4 BP180
LL
Laminin-5
LD
AF
AP
A
Intact skin
Epidermis
LL
LD
B
epidermolysis is somewhat illogical because epidermal disruption is not the primary change in several categories of EB. Nevertheless, the name epidermolysis bullosa, as originally used by Koebner in 1886,2 is now so well established in the literature that it remains the accepted term. All forms of EB are rare; it is estimated that globally about 500 000 people have EB.
Dermis
NaCl-split skin
Epidermis
Artificial blister cavity
Classification of EB Historically, the classification of EB has been difficult and not helped by the large variety of names and eponyms that have traditionally been used. While these early observations were important in establishing EB as an entity, a major step forward was made by Pearson in the 1960s,3 who used transmission electron microscopy to show that the ultrastructural level of tissue cleavage (blister formation) in the skin is distinctive in the three major groups of EB: intraepidermal in EB simplex, through the lamina lucida in junctional EB, and below the lamina densa in dystrophic EB (although a separate fourth category, Kindler syndrome, has subsequently been added in which there is a variable mixed level of blistering).4 Indeed, these different levels of cleavage still form the basis of the current classification of EB (Table 4.1).1 Since Pearson’s initial observations, the concept and content
LD
Dermis
of EB simplex has also expanded with the addition of more superficial skin fragility disorders, including inherited disorders of desmosomes as well as cornification (Table 4.2). Likewise, the classification of the junctional and dystrophic forms of EB has also evolved, although the multiple variants thereof have largely been defined by clinical features (Tables 4.3 and 4.4). Although the clinicopathologic entities now regarded as forms of EB have expanded, most of the major forms of EB occur at or close to the dermal– epidermal junction, and a schematic of the key proteins and genes implicated in these forms of EB is shown in Fig. 4.9.
120 Inherited and autoimmune subepidermal blistering diseases
A
B
121 Epidermolysis bullosa
Level of skin cleavage EB type EB subtype Defective protein(s)
Intraepidermal EB simplex Suprabasal EB simplex Transglutaminase-5; Plakophilin-1; Desmoplakin; Plakoglobin Basal EB simplex Keratins 5 and 14; Plectin; Exophilin-5; 230-kDa bullous
pemphigoid antigen/dystonin
Intralamina lucida Junctional EB Junctional EB generalized Laminin-332; Type XVII collagen α6, β4, α3 integrin subunits Junctional EB localized Type XVII collagen; Laminin-332; α6, β4 integrin subunits
Sublamina densa Dystrophic EB Dominant Dystrophic EB Type VII collagen Recessive Dystrophic EB Type VII collagen
Mixed Kindler syndrome Kindlin-1 (Fermitin family homologue-1)
EB simplex type EB simplex subtype Targeted protein(s)
Suprabasal Acral peeling skin syndrome Transglutaminase-5 EB simplex superficialis Unknown Ectodermal dysplasia-skin
Plakophilin-1
fragility syndrome
Severe acantholytic EB Desmoplakina,
Dystrophic EB typea Dystrophic EB subtype
Dominant Generalized Acral Pretibial Pruriginosa Nails only Bullous dermolysis of the newborn
Plakoglobina
Basal EB simplex – localizedb Keratins 5 and 14 EB simplex – generalized
Keratins 5 and 14
severe
EB simplex – generalized
Keratins 5 and 14
intermediate
EB simplex – mottled
Keratin 5
pigmentation
EB simplex – migratory
Keratin 5
circinate
EB simplex – autosomal
Keratin 14
recessive (K14)
EB simplex – autosomal
230 kDa Bullous
recessive (BP230)
pemphigoid antigen EB simplex – autosomal
Exophilin-5
Recessive Generalized severe Generalized intermediate Inversa Localized Pretibial Pruriginosa Centripetalis Bullous dermolysis of the newborn
aThe targeted protein in all types and subtypes of dystrophic EB is type VII collagen.
In addition to establishing specific levels of blistering in the different subtypes of EB, ultrastructural studies were also able to identify distinct morphologic abnormalities, such as keratin filament disruption in EB simplex, poorly formed hemidesmosomes in junctional EB, and rudimentary anchoring fibrils in dystrophic EB. During the 1980s, the immunohistochemical labeling of EB skin with basement membrane zone antibodies became a useful diagnostic addition, for example showing reduced immunostaining for proteins such as laminin-332 in some forms of junctional EB, and type VII collagen in recessive dystrophic EB, respectively. Then in the 1990s, the discovery of candidate genes and pathogenic mutations, such as mutations in KRT14 and KRT5 in EB simplex (keratins 14 and 5) and COL7A1 in dystrophic EB (type VII collagen), heralded the era of molecular diagnostics. Reflecting these advances at electron microscopic, immunohistochemical, and molecular levels, the classification of EB has evolved over the years with the most recent international consensus report (published in 2014) recommending abandonment of most historical eponyms and adoption of an “onion skin” approach to disease nomenclature – the different layers referring to the methods of diagnostic evaluation available – clinical phenotype, mode of transmission, level of split in the skin, immunohistochemistry, mutation detection, etc.1 For example, a patient with intraepidermal blister formation, blistering confined to the palms and soles, and a family history consistent with autosomal dominant transmission would initially be classified as having localized EB simplex. Once mutational confirmation is completed, the final diagnosis, using the onion skin method, would be: EB simplex, localized, KRT5 mutation (missense mutation). It is believed that this format for diagnosing EB provides both practical and academic value in classifying EB, although it is likely to be revised further in years to come as more discoveries are made.
recessive (exophilin-5)
EB simplex – muscular
Plectin
dystrophy
Plectin; α6, β4 integrin
EB simplex – muscular
dystrophy
subunits EB simplex – Ogna Plectin
aAlthough listed as being associated with suprabasal subtypes of EB simplex, desmoplakin and plakoglobin show that pan-epidermal expression and mutations in these proteins affect basal as well as suprabasal keratinocytes. bOccasionally, this type of EB simplex can also result from mutations in plectin, type XVII collagen and the β4 integrin subunit.
Junctional EB type Junctional EB subtype Targeted protein(s)
Generalized Severe Laminin-332 Intermediate Laminin-332; Type XVII
collagen Late onset Type XVII collagen With pyloric atresia α6 and β4 integrin
subunits With respiratory and
α3 integrin subunit
renal involvement
Localized Localized Laminin-332; Type XVII
collagen, α6 and β4 integrin subunits Inversa Laminin-332 Laryngo-onycho-
Laminin α3a
cutaneous syndrome
Molecular studies have now identified pathogenic mutations in 18 different genes in the heterogeneous clinical subtypes of EB (Table 4.5).5 With regard to diagnosing EB, sequencing of patient DNA (either Sanger sequencing or next generation sequencing using whole exome sequencing or selected
122 Inherited and autoimmune subepidermal blistering diseases
230-kDa
BULLOUS PEMPHIGOID ANTIGEN
PLECTIN
Keratin 5: KRT5
KERATINS
EB simplex (AD)
5 and 14
Dowling-Degos disease (AD)
Keratin 14: KRT14
EB simplex (AD and AR)
Naegeli-Franceschetti-Jadassohn syndrome (AD)
Plectin: PLEC1
Hemidesmosome
β
4 INTEGRIN
EB simplex-muscular dystrophy (AR) EB simplex-pyloric atresia (AR) EB simplex (AD and AR)
230-kDa bullous pemphigoid antigen: BPAG1
EB simplex (AR)
Collagen XVII: COL17A1
Generalized intermediate junctional EB (AR and AD)
COLLAGEN XVII CD 151
Lamina Lucida
a6 integrin: ITGA6
Junctional EB-pyloric atresia (AR)
EB simplex-pyloric atresia (AR)
b
4 integrin: ITGB4
α
Lamina Densa
LAMININ 332
Dermis
COLLAGEN VII
Junctional EB-pyloric atresia (AR)
6 INTEGRIN
EB simplex-pyloric atresia (AR) Generalized intermediate junctional EB (AR)
EB simplex (AR)
Laminin-332: LAMA3, LAMB3, LAMC2
Generalized intermediate junctional EB (AR) Generalized intermediate severe EB (AR)
Laryngo-onycho-cutaneous syndrome (LAMA3, AR)
Collagen VII: COL7A1
Dystrophic EB (AD and AR)
gene panels) is gradually emerging as a primary investigation with the role of skin biopsy somewhat diminishing. This is certainly true for autosomal dominant forms of EB simplex or dominant dystrophic EB in which the skin pathology (both ultrastructural and immunohistochemical) may not differ significantly from normal skin, especially if no fresh blister is included in the biopsy. Nevertheless, skin biopsy remains a useful means of rapidly diagnosing autosomal recessive forms of EB, which is particularly useful in neonates with fragile skin since prompt diagnosis has important implications for optimal clinical management of affected babies and their families. Details about the most appropriate skin biopsy techniques and investigations for diagnosing EB are presented in Chapter 2.
therefore work suboptimally (or not at all) on paraffin-embedded skin sections. For EB diagnostics, skin immunohistochemistry is best performed on frozen sections (see Chapter 2). Part of the diagnostic skin biopsy is often fixed and processed for transmission electron microscopy although oftentimes sections are only cut if the skin immunolabeling fails to establish a diagnosis. Over the last decade, therefore, with the increasing utility of antibodies to skin basement membrane and epithelial proteins, the number of EB skin biopsies requiring transmission electron microscopy to establish a diagnosis has reduced by over 50%. This number is likely to decrease further as molecular diagnostics become more widely available, cheaper and quicker. Immunoelectron microscopy is not performed for routine EB diagnostics and is mainly used for research, including translational research when evaluating clinical trials of cell and gene-based therapies.
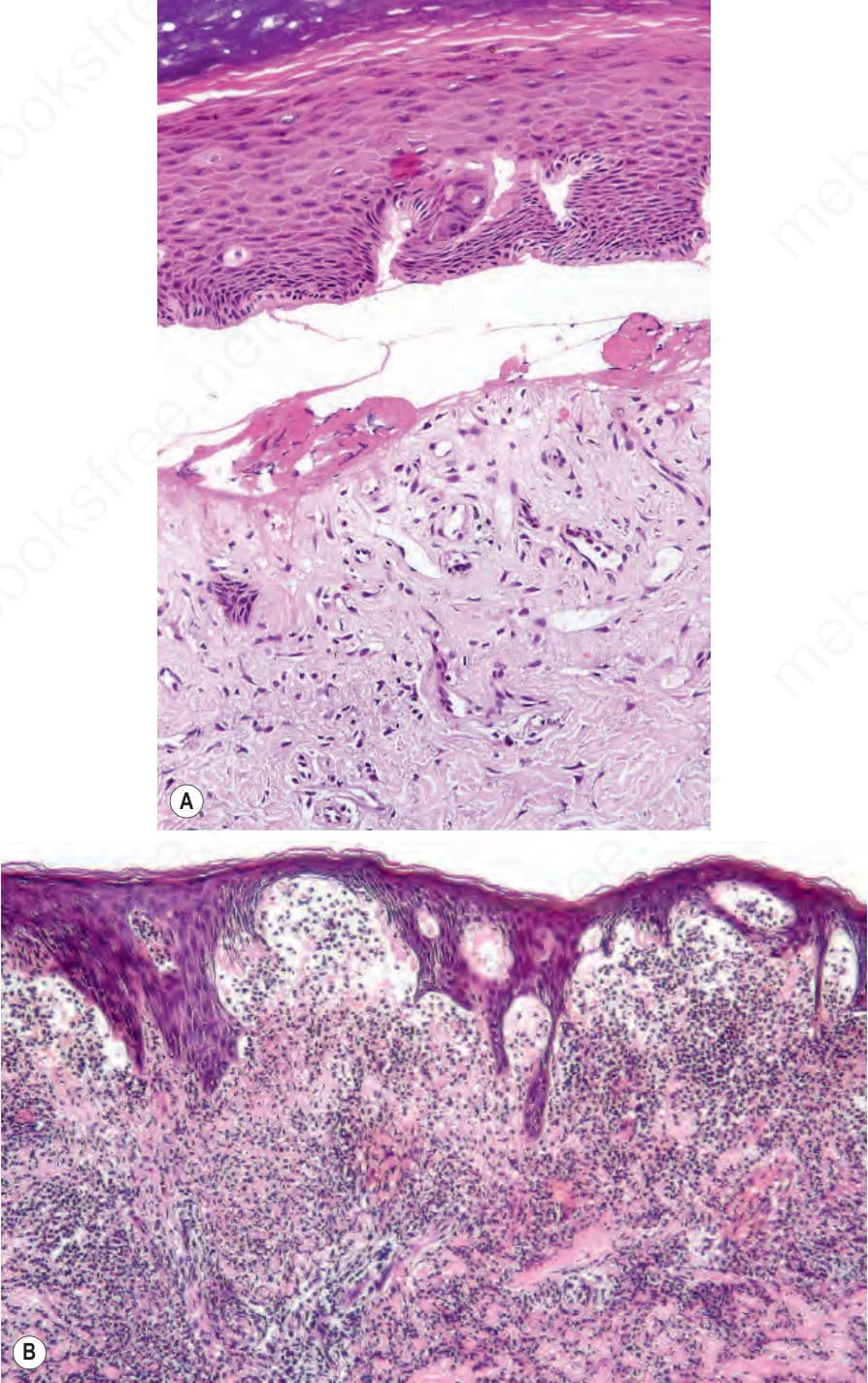
Fig. 4.1 Classification of subepidermal blisters: lesions may be subdivided into (A) cellpoor and (B) cell-rich variants.
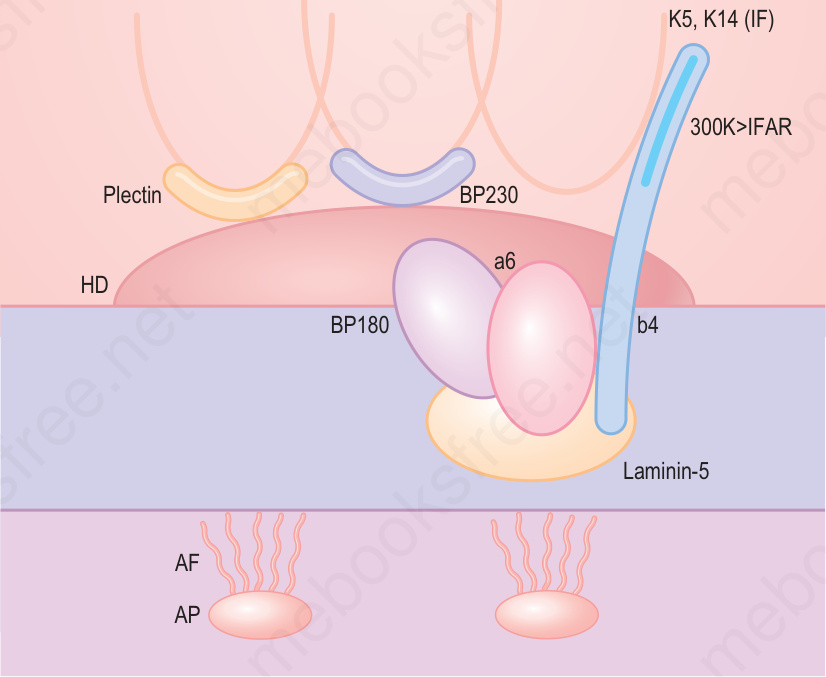
Fig. 4.2 Basement membrane constituents: blisters can be classified into those that develop within the lamina lucida (LL) and those that arise below the lamina densa (LD). (AF, anchoring fibrils; AP, anchoring plaque; CM, cell membrane.)
Fig. 4.3 Split skin immunofluorescence.
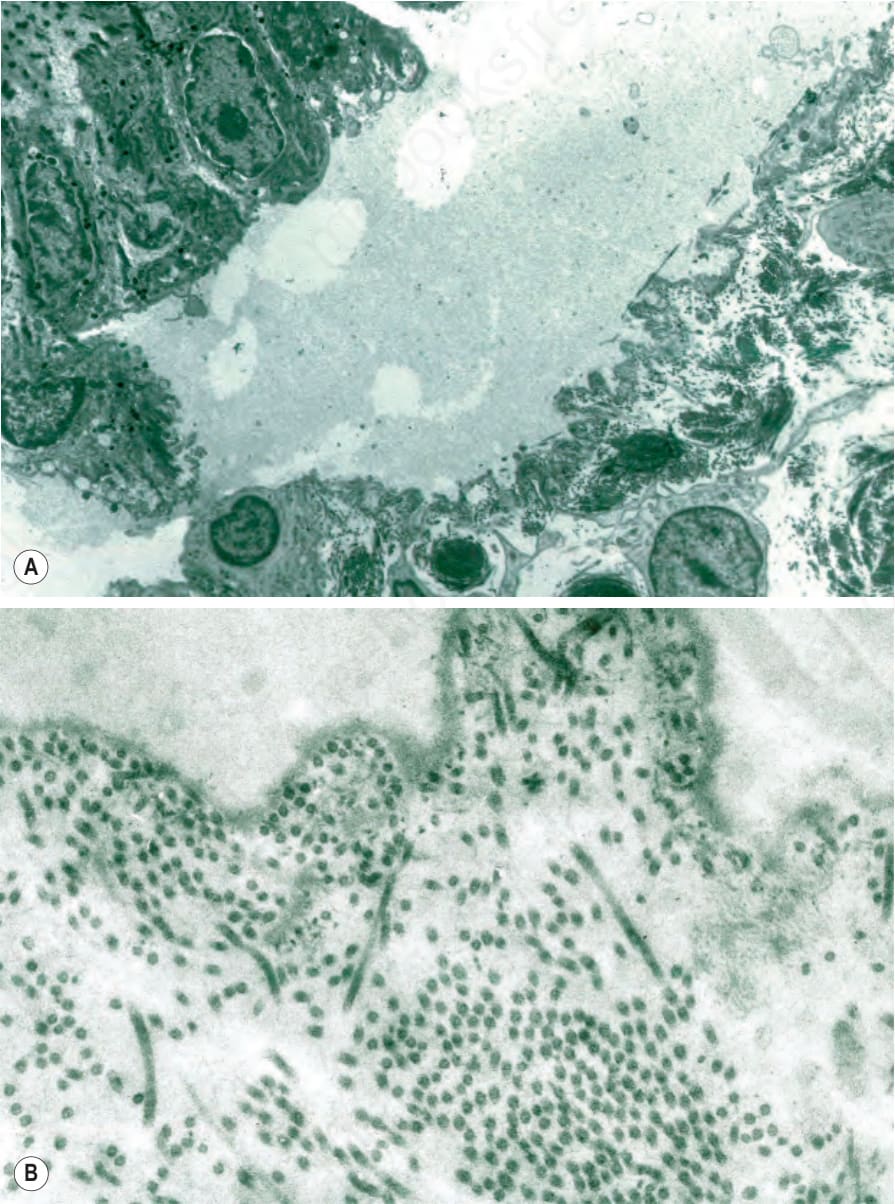
Fig. 4.4 (A, B) Split skin immunofluorescence: the split is through the lamina lucida, the lamina densa lining the floor of the artificial blister cavity.
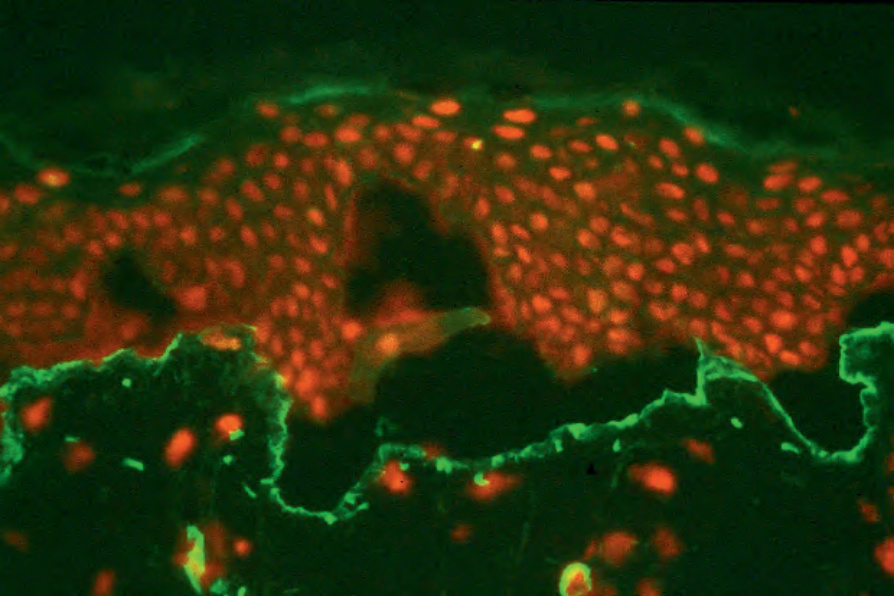
Fig. 4.5 Split skin immunofluorescence: type IV collagen lines the floor of the split skin artificial blister which therefore forms within the lamina lucida. By courtesy of B. Bhogal, FIMLS, Institute of Dermatology, London, UK.
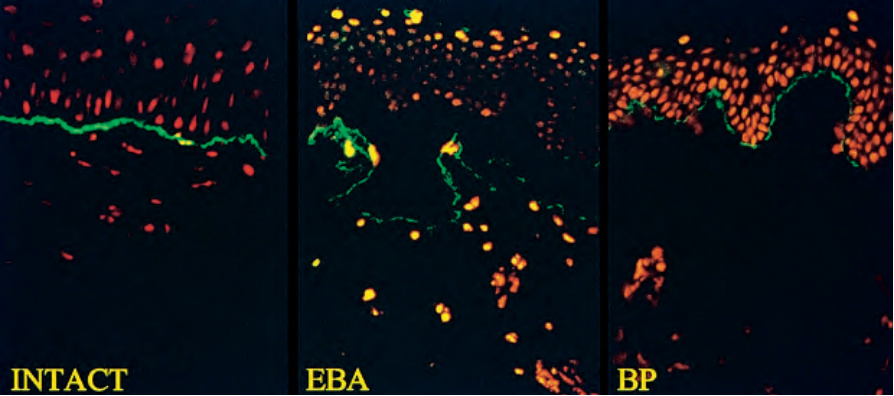
Fig. 4.6 Split skin immunofluorescence: (left) linear IgG at the basement membrane; (middle) in epidermolysis bullosa acquisita (EBA), the antibody binds to the floor of the blister cavity; (right) in bullous pemphigoid (BP), the antibody binds to the roof of the blister. By courtesy of B. Bhogal, FIMLS, Institute of Dermatology, London, UK.
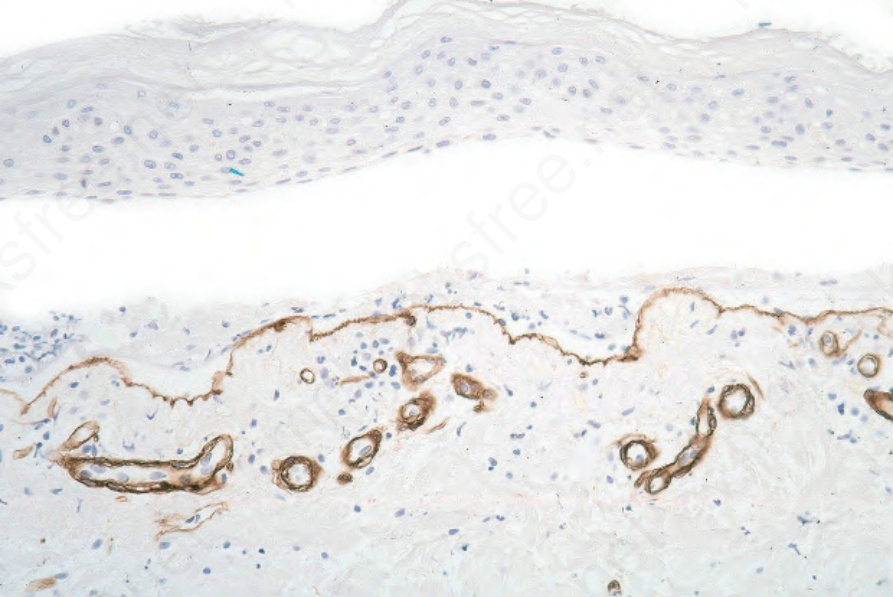
Fig. 4.7 Paraffin-embedded immunoperoxidase antigen mapping: in bullous pemphigoid, type IV collagen is present along the floor of the blister.
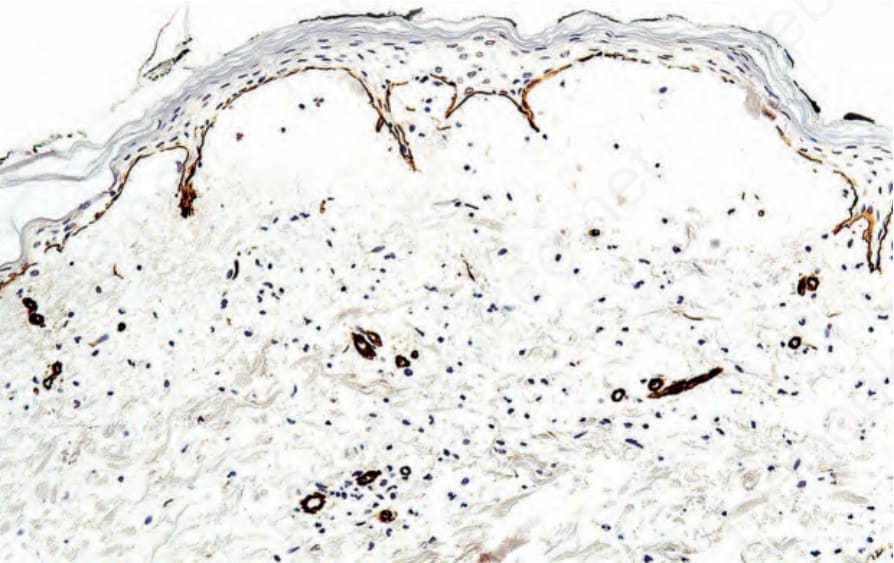
Fig. 4.8 Paraffin-embedded immunoperoxidase antigen mapping: in epidermolysis bullosa acquisita, type IV collagen is present along the roof of the blister cavity.
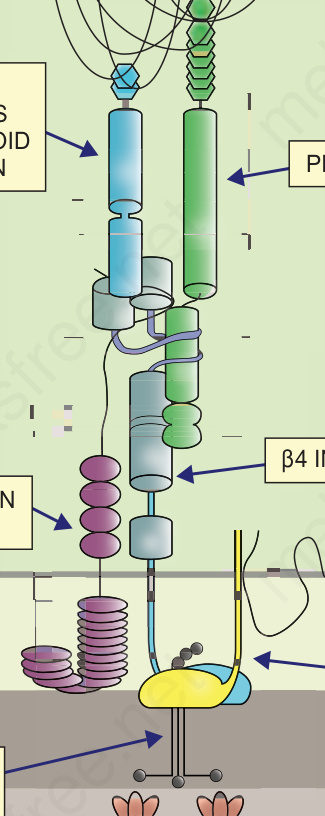
Fig. 4.9 Schematic representation of the major adhesive proteins within hemidesmosome adhesion complexes at the dermal–epidermal junction and their involvement in different types of EB. (AD = autosomal dominant; AR = autosomal recessive).
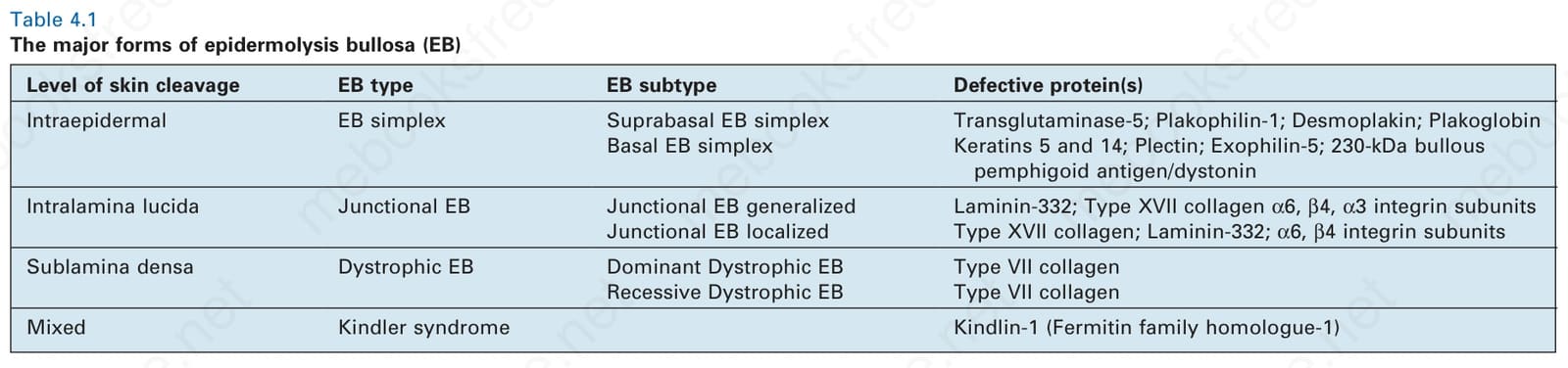
Table 4.1 The major forms of epidermolysis bullosa (EB)
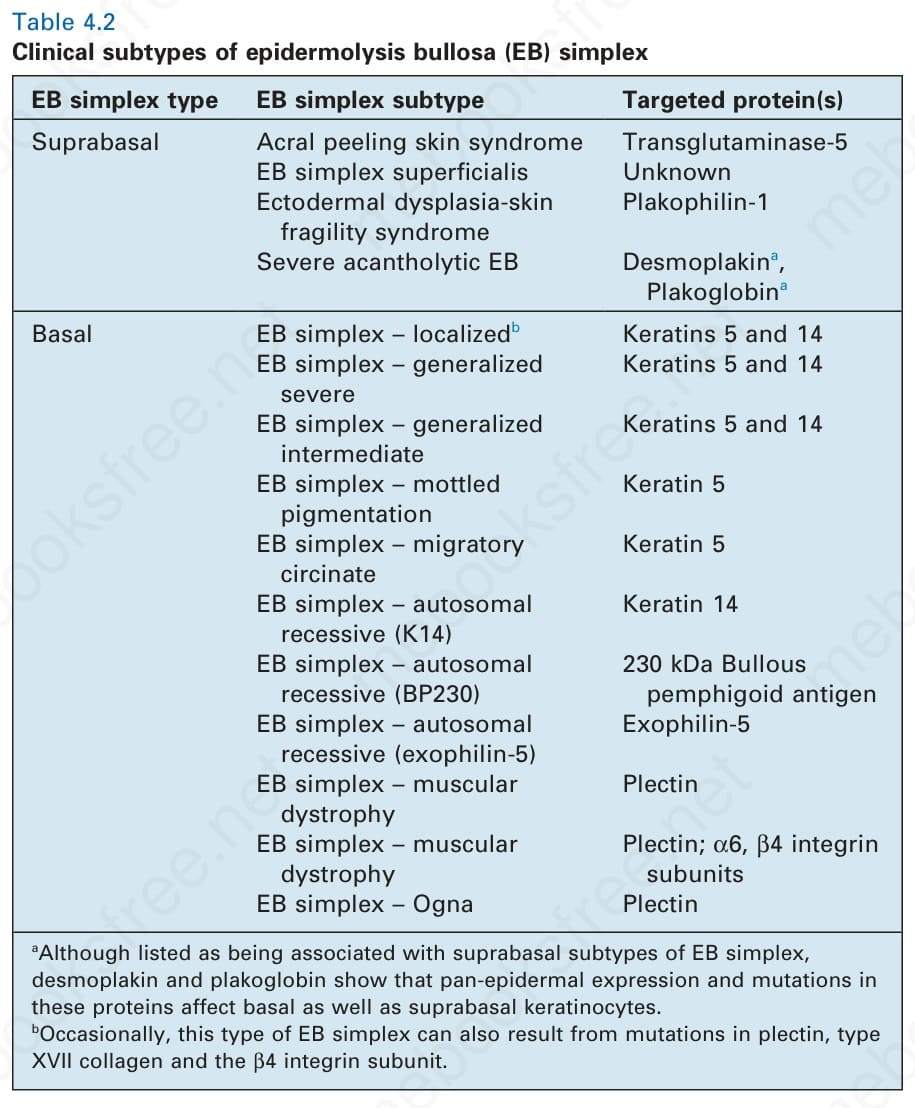
Table 4.2 Clinical subtypes of epidermolysis bullosa (EB) simplex
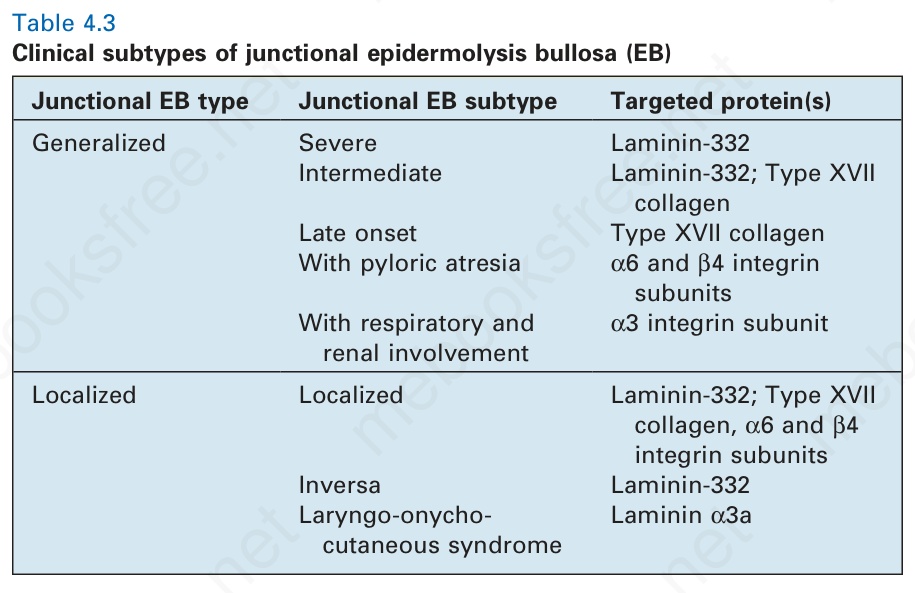
Table 4.3 Clinical subtypes of junctional epidermolysis bullosa (EB)
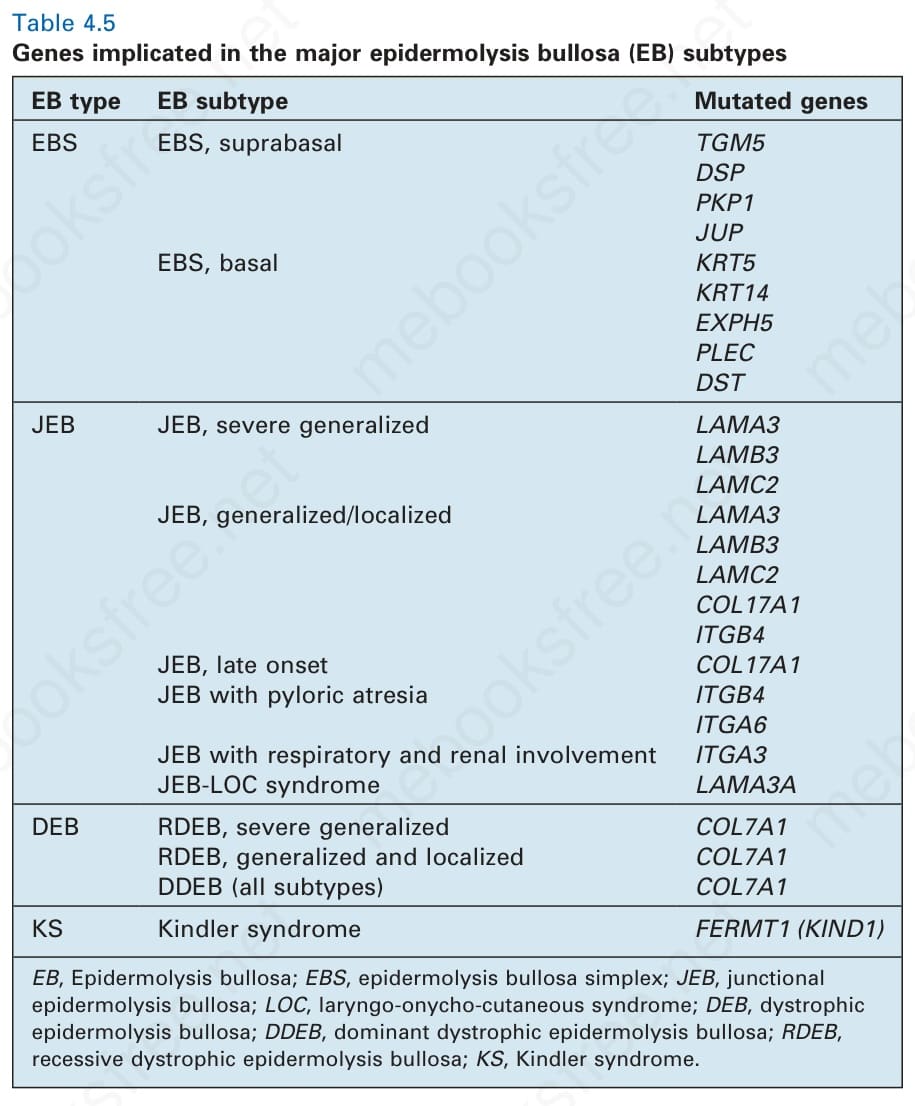
Table 4.5 Genes implicated in the major epidermolysis bullosa (EB) subtypes
For the dermatopathologist, a key issue will always be whether EB is best classified by skin/molecular pathology or by clinical morphology, and therefore in the sections that follow, both options are presented. Another important issue is how best to diagnose EB with the material made available. For skin that has been formalin-fixed and embedded in paraffin, successful diagnosis may depend on the presence or absence of a fresh blister. Most clinicians are aware that biopsying a blister that has been present for more than 12 hours is likely to present the pathologist with difficulties in determining the true plane of cleavage because of re-epithelialization and therefore rubbed non-blistered skin is preferentially sampled (see Chapter 2). Antigen mapping and immunoperoxidase staining is possible with antibodies to type IV collagen, which determines whether the lamina densa is present in the roof (dystrophic EB) or base (EB simplex and junctional EB) of the blister. Immunodiagnosis is also possible using selective probes, such as type VII collagen (reduced in recessive dystrophic EB). Nevertheless, the majority of the antibodies used to diagnose EB target transmembranous proteins and
Clinical features of EB EB simplex The subtypes of EB simplex are subdivided into basal and suprabasal variants, that manifest as at least 15 distinct clinical disorders (Table 4.2).
Localized EB simplex This is the most common type of EB.6 Inheritance is autosomal dominant. The soles and palms (Fig. 4.10) are mainly affected. The condition is typically worse in warm weather. Hyperhidrosis of the feet is common; this increases friction, which also exacerbates blistering. The blisters usually heal without scarring or milia formation. Calluses are very common, especially in adults. Oral blistering occurs in less than 25% of cases. The hair and teeth are normal.
123 Epidermolysis bullosa
EB type EB subtype Mutated genes
EBS EBS, suprabasal TGM5 DSP PKP1 JUP EBS, basal KRT5 KRT14 EXPH5 PLEC DST
JEB JEB, severe generalized LAMA3 LAMB3 LAMC2 JEB, generalized/localized LAMA3 LAMB3 LAMC2 COL17A1 ITGB4 JEB, late onset COL17A1 JEB with pyloric atresia ITGB4 ITGA6 JEB with respiratory and renal involvement ITGA3 JEB-LOC syndrome LAMA3A
DEB RDEB, severe generalized COL7A1 RDEB, generalized and localized COL7A1 DDEB (all subtypes) COL7A1
KS Kindler syndrome FERMT1 (KIND1)
EB, Epidermolysis bullosa; EBS, epidermolysis bullosa simplex; JEB, junctional epidermolysis bullosa; LOC, laryngo-onycho-cutaneous syndrome; DEB, dystrophic epidermolysis bullosa; DDEB, dominant dystrophic epidermolysis bullosa; RDEB, recessive dystrophic epidermolysis bullosa; KS, Kindler syndrome.
Acral peeling skin syndrome
A differential diagnosis of localized autosomal dominant EB simplex is autosomal recessive acral peeling skin syndrome, although both are classified as forms of EB simplex.7 Blisters typically occur on the feet but are often more evident on the sides or dorsal aspects of the toes (Fig. 4.11). The level of blistering occurs above the granular layer, but because of the thicker stratum corneum in acral skin the clinical consequences may be almost identical to blistering through the basal keratinocyte layer.
Generalized severe EB simplex A further common subtype of EB simplex is the generalized severe variant, previously known as Dowling-Meara EB simplex.8 Inheritance is autosomal dominant. Blisters tend to occur in groups, hence the earlier use of the term EB herpetiformis (Fig. 4.12). In infancy, blistering may be severe with involvement of the mucous membranes, shedding of nails, and formation of milia. The distinctive feature is spontaneous herpetiform, annular or arcuate blistering on the trunk, limbs, and neck. Irregular hyperkeratosis of the palms and soles or keratoderma is often present. The general condition tends to improve with age.
Generalized intermediate EB simplex This subtype of EB includes inherited blistering previously known as Koebner EB simplex.9 Most cases are autosomal dominant. Although
124 Inherited and autoimmune subepidermal blistering diseases
EB simplex autosomal recessive keratin 14 Although much less common than autosomal dominant disease, recessive mutations in keratin 14 cases may resemble dominant generalized intermediate EB simplex.13 Blisters are often scattered and there is usually minor palmoplantar keratoderma and varying degrees of nail dystrophy, atrophic scarring, hyperpigmentation, and oral and genital blistering. There is often a complete loss of keratin 14 expression in the epidermis, although the phenotype is typically a less severe phenotype than some dominant cases. No recessive cases involving keratin 5 have yet been reported.
EB simplex with muscular dystrophy Inherited skin fragility can occur in association with muscular dystrophy, although myasthenia gravis and spinal muscular atrophy have also been described.14 The blisters, which affect the skin and oral mucosa, are present at birth or soon afterwards. Muscle weakness and wasting may be severe and evident in early childhood, or milder and only detectable later in life. The blistering may be widespread, but can be limited to the hands and feet. There can be atrophic scarring, milia, nail dystrophy, and alopecia. Supraglottic scarring and hoarseness may necessitate tracheostomy. The disorder is autosomal recessive and results from mutations in plectin.
EB simplex with pyloric atresia Pyloric atresia may rarely occur in the setting of EB simplex – most cases of which are classified as a junctional form of EB.15 Affected infants tend to have generalized skin disease. Although only a few cases have yet been reported, this entity appears to be as severe as junctional EB with pyloric atresia and clinically impossible to differentiate from it. There are widespread blisters and erosions that increase infection risk with early demise in the neonatal period. Inheritance is autosomal recessive and involves mutations in plectin.
usually mild, approximately 60% of patients have localized scarring and approximately 15% have milia. The development of hair, teeth, and nails is normal. Blisters appear within the first year and may be present at birth. In infancy, they commonly appear on the occiput, back, and legs, while in childhood the hands and feet are often affected, although the palms and soles are not preferentially involved, as in localized EB simplex. In common with other forms of EB simplex, blistering is worse in warm weather.
EB simplex autosomal recessive BP230 Mutations leading to a complete loss of the 230-kDa bullous pemphigoid antigen results in a rare form of autosomal recessive of EB simplex.16 Blistering is lifelong and generalized but clinically is mild with only a few predominantly acral blisters that can extend to several centimeters in size, but which are noninflammatory and which heal with no scarring and only mild postinflammatory pigmentary anomalies. Oral and genital mucosae are not affected.
EB simplex with mottled pigmentation Distinction from other forms of EB simplex is made by the presence of pigmentary changes, which are present at birth or appear during infancy.10 There is a reticulate pattern of small, tan-colored macular lesions, which may spread from acral sites to the trunk and which fade with age (Fig. 4.13). They may cover the entire skin surface, but preferentially involve the neck, upper trunk, or extremities. Blistering may be localized, or become more generalized. Most cases result from a specific heterozygous missense mutation in keratin 5.
EB simplex autosomal recessive exophilin-5 Autosomal recessive loss-of-function mutations in EXPH5 (encoding exophilin-5, also known as Slac2-b) result in mild, scattered, trauma-induced skin fragility, although blistering can be generalized shortly after birth.17 In later infancy, however, the clinical features mostly comprise crusted erosions on the limbs with few intact blisters. No mucosal abnormalities are noted. Typically, the disease severity improves with age and may even remit completely.
EB simplex plakophilin-1 deficiency This disorder represents the first inherited disorder of desmosomes; it is also known as ectodermal dysplasia skin fragility syndrome.18 Inheritance is autosomal recessive. Widespread erosions are present at birth, with evident perioral cracking and hypotrichosis. During infancy, the skin manifestations evolve to reveal trauma-induced scale crusting, a prominent palmoplantar keratoderma with painful fissures, and nail dystrophy. Most cases have substantial loss of scalp hair and eyebrows and the perioral changes persist. Unlike some other inherited desmosomal disorders, there is no cardiac involvement in this condition since plakophilin-1 is not expressed in the heart.
EB simplex Ogna This autosomal dominant condition was named after the village in Norway where the first affected family originated.11 There is seasonal blistering of the hands and feet, and occasionally elsewhere. This rare subtype of EB is distinguishable clinically by a generalized bruising tendency, hemorrhagic bullae, and toenail dystrophy. The disorder results from a particular missense mutation in plectin that renders the protein more susceptible to proteolysis, thus reducing its expression and function.
EB simplex migratory circinate This is a rare form of autosomal dominant EB simplex that is characterized by migratory circinate erythema and post-inflammatory hyperpigmentation.12 Present at birth, blistering is generalized. The underlying cause is an atypical mutation in keratin 5 that leads to skin inflammation with a T-cell infiltrate.
EB simplex desmoplakin deficiency Autosomal recessive mutations in the desmosomal plaque protein, desmoplakin, can result in a very severe form of inherited skin fragility. It was originally termed ‘lethal acantholytic EB,’19 although the latest classification of EB has tried to avoid using emotive terms such as ‘lethal’ given the impact such a devastating term can have on families as well as the possible
unpredictability in disease course in some cases. Blistering is present at birth and generalized. A characteristic feature is the presence of oozing erosions rather than frank blisters. Other findings include markedly abnormal nails, neonatal teeth, intraoral erosions, and alopecia. The gastro-intestinal, genito-urinary, and respiratory tracts may also be involved.
EB simplex plakoglobin deficiency Autosomal recessive mutations in plakoglobin, a protein found in desmosomal plaques and adherens junctions, can also result in a very severe form of inherited skin fragility. This was originally termed ‘lethal congenital EB,’20 although, as is the case for desmoplakin, the latest consensus on EB classification has avoided use of the word ‘lethal’ preferring to opt for skin fragility plakoglobin deficiency (or severe acantholytic EB) instead. Clinically there are widespread erosions and massive transcutaneous fluid loss leading to a poor prognosis. Cardiac involvement is possible with some inherited abnormalities of plakoglobin.
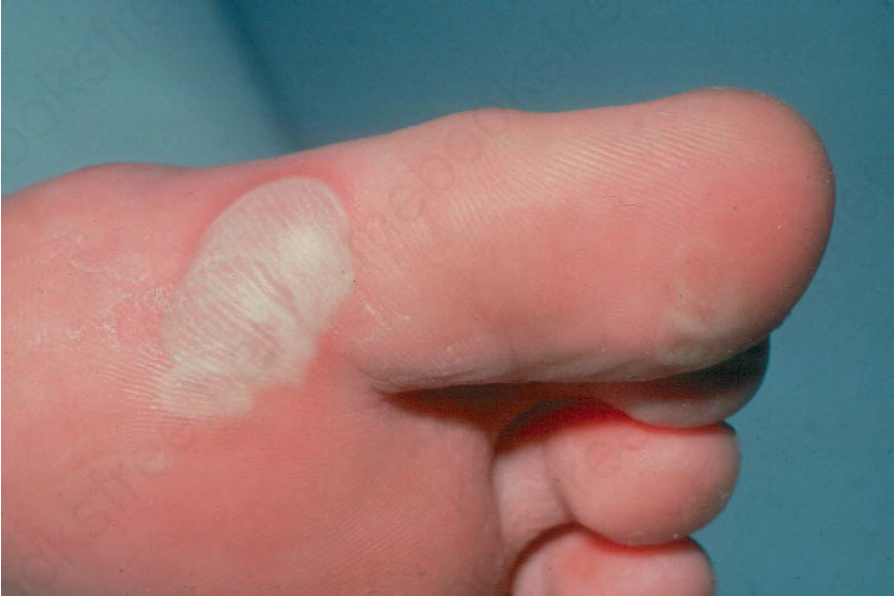
Fig. 4.10 Localized EB simplex: typical acral lesions affecting the toes. The pale color is due to the marked thickness of the roof of the blister. By courtesy of St John’s Institute of Dermatology, London, UK.
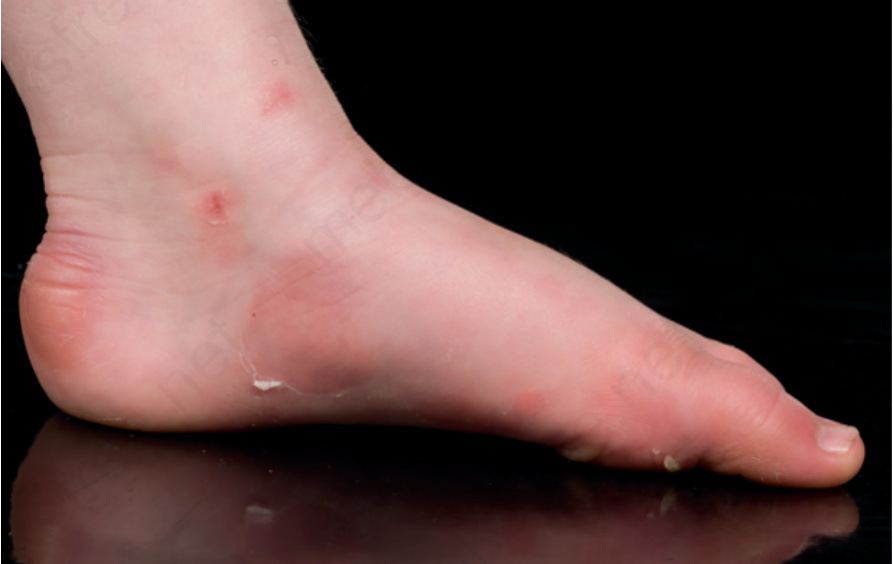
Fig. 4.11 Acral peeling skin syndrome: signs of peeling and erythema on the ankle and instep extending to the toes.
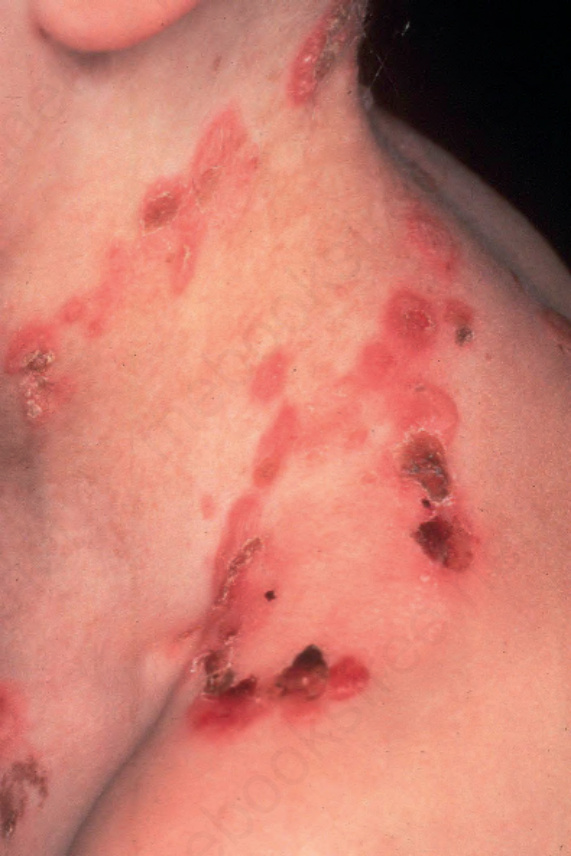
Fig. 4.12 Generalized severe EB simplex: showing characteristic grouping of blisters and erosions. By courtesy of R.A.J. Eady, MD, St John’s Institute of Dermatology, London, UK.
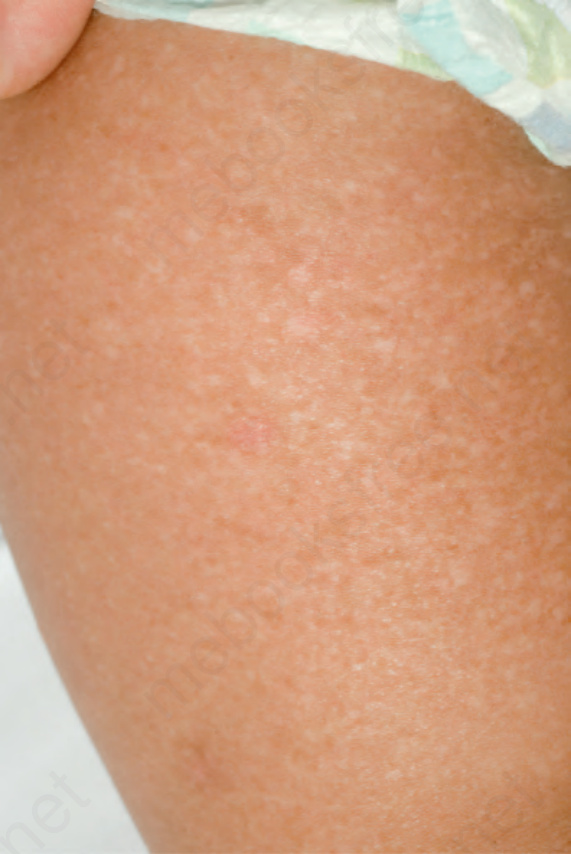
Fig. 4.13 EB simplex with mottled pigmentation: diffuse hyper- and hypopigmentation with minor small patches of erythema and occasional tiny vesicles. By courtesy of J.E. Mellerio, St John’s Institute of Dermatology, London, UK
125 Epidermolysis bullosa
EB simplex superficialis This autosomal dominant condition is characterized by the presence of superficial erosions rather than intact blisters, similar to those seen in pemphigus foliaceus.21 Epidermal cleavage is typically just beneath the stratum corneum. Healing results in localized atrophic scarring or postinflammatory hyperpigmentation. The molecular basis of this subtype of EB simplex is unknown and therefore it is uncertain whether this condition is truly a discreet clinicopathological entity.
Junctional EB Almost all clinical variants of junctional EB are characterized by autosomal recessive inheritance and by blister formation at the level of the lamina lucida. Conventionally, junctional EB is divided into two main categories: generalized and localized, each of which has a number of subtypes (Table 4.3). The terms Herlitz, non-Herlitz, generalized atrophic benign, cicatricial, progressive, and atrophicans are no longer used in the updated classification of EB, nor are the words ‘lethal’ or ‘letalis’ considered appropriate diagnostic labels.
Generalized severe junctional EB This is the most severe form of junctional EB. Blistering and erosions are present at or soon after birth and rapidly become generalized.22 The whole skin is fragile and moving the baby may cause extensive blistering (Fig. 4.14). Nail bed involvement is very common (Fig. 4.15). Eroded areas are often very slow to heal and may lead to atrophic scarring. Milia are not generally seen. Involvement of the oral and pharyngeal mucosa is frequent and may be severe; hoarseness and stridor may indicate laryngeal or supraglottic involvement, most notably potentially life-threatening stenosis or stricture. Many infants die early in infancy with overwhelming infection or from failure to thrive, but those surviving the first few months will often develop distinctive lesions characterized by non-healing, crusted erosions containing exuberant granulation tissue.
Generalized intermediate junctional EB The early clinical course of this subtype of junctional EB may be similar to the generalized severe variant, but the patients usually survive to adulthood.23 There is a gradual lessening in blistering severity with age. Mucous membranes are involved, and the teeth show severe enamel defects or may fail to erupt normally. Nails are often dystrophic or missing. Atrophic scarring is characteristic. There is also alopecia affecting the scalp, eyebrows, and eyelashes, and body hair is also sparse or absent. Large pigmented nevi, or acquired macular hyperpigmented lesions, are typical. In this type of junctional EB, it is common to observe small patches of skin that do not blister; this phenomenon is called revertant mosaicism (also known as natural gene therapy), and represents a spontaneous correction of one copy of the mutant gene.
Generalized late-onset junctional EB This is a rare subtype of autosomal recessive junctional EB that overlaps with generalized intermediate junctional EB, except that the onset of
symptoms is delayed – often not starting until childhood, typically between 5 and 8 years of age.24 Initially, the trauma-induced blisters mainly occur on the hands and feet, although they may be preceded by nail dystrophy. Later, knees and elbows are involved. Progressive atrophic changes lead to early loss of fingerprint patterns and mild finger contractures. The tooth enamel may be defective and the tongue papillae may disappear. The oral mucosa is sometimes involved.
Junctional EB with pyloric atresia Blistering is usually present at birth, following a pregnancy complicated by polyhydramnios. The lesions are usually widespread and can result in atrophic scarring (Fig. 4.16).25 The teeth are hypoplastic, lacking normal
126 Inherited and autoimmune subepidermal blistering diseases
A
enamel, and the nails are dystrophic. Early attempts at feeding result in vomiting. The majority of cases do not survive early infancy. In those children who survive, other features include haematuria, dysuria, and recurrent urinary tract infections. The pyloric canal is often completely obliterated (Fig. 4.17) and requires surgical correction although the nature of the underlying molecular pathology usually influences prognosis.
Junctional EB with respiratory and renal involvement This is a recent addition to the classification of junctional EB. The features comprise congenital nephrotic syndrome, interstitial lung disease, and skin fragility, due to autosomal recessive mutations in α3 integrin.26 The renal and respiratory features predominate clinically, and skin blistering may be a minor feature that only occurs later. The oral mucosa is not involved. The scalp hair, eyebrows, and eyelashes are fine and sparse and nails may be dystrophic. Prognosis is poor with recurrent lung infections and multiorgan failure consistent with the known distribution of α3 integrin in several tissues.
Localized junctional EB Localized forms of junctional EB occur: typical clinical manifestations include nail dystrophy, dental enamel changes, and blistering involving the lower legs and feet only.27 In some individuals, chronic, painful erosions associated with hyperkeratosis develop on the soles, although it is not clear why the lower legs should be a predilection site for blistering. In some cases, blistering starts in neonates, while in others there may be late-onset disease.
Localized junctional EB inversa In the neonatal period in the rare inversa subtype of localized junctional EB, the whole skin may be fragile with generalized blistering. Later, however, the lesions affect chiefly the groins, perineum, and axillae, hence the description of ‘inversa.’28 Healing may result in small, atrophic white streaks. Dysplastic teeth, erosions of the cornea, and feet and nail dystrophy are all features. Why there should be a preference for flexural site involvement is not known.
B
Dystrophic epidermolysis bullosa Dystrophic EB can be autosomal recessive or autosomal dominant (Table 4.4). Clinically, dystrophic EB is characterized by skin fragility, blistering, scarring, nail dystrophy, and milia formation. Mucosal involvement is common and erosions and scarring can affect the mouth, esophagus, genitalia, and anus. There may be clinical overlap between some cases of recessive and dominant dystrophic EB, which can make genetic counseling difficult, particularly in sporadic cases. The most recent classification of EB no longer contains any eponymous subtypes, and also recognizes that the phenotypic appearances vary considerably between patients and that, given the spectrum of clinical appearances, diagnostic labeling within an individual category can be somewhat arbitrary in some cases.
Localized junctional EB laryngo-onycho-cutaneous syndrome This subtype of junctional EB starts in infancy with chronic erosions affecting the face (mainly around the nose and mouth) although erosions are also seen on the limbs, trunk, and genitalia.29 The nails are also involved with marked periungual and subungual inflammation and a universal feature is hoarseness. The teeth may be notched. There is prominent skin and mucosal granulation tissue that can lead to delayed wound healing, laryngeal obstruction, and blindness. The disorder results from autosomal recessive mutations in a particular splice variant of the laminin α3 polypeptide.
Dominant dystrophic EB generalized Blisters in dominant dystrophic EB mainly occur following trauma to the skin overlying the bony prominences, such as the knees and ankles, and dorsa of the hands or feet (Fig. 4.18).30 The most consistent findings are localized scarring with milia formation and dystrophic nails. Nail dystrophy is probably the most important diagnostic feature of the disease, especially in adults, because many patients have only limited blistering and scarring, which becomes less noticeable with age. Blistering in the mouth is usually mild and the teeth are generally normal. However, perianal lesions may cause considerable pain, especially in children. Terms such as Bart syndrome are obsolete. Cutaneous features, such as albopapuloid lesions, once
127 Epidermolysis bullosa
pruriginosa (see next section). Clinical signs are not exclusively confined to the shins and relatively minor skin fragility, scarring, and milia may be detected at other body sites, particularly over bony prominences.
Dominant or recessive dystrophic EB pruriginosa This subtype of dystrophic EB overlaps clinically with the pretibial variant. The main difference is the intense pruritus, the etiology of which is uncertain.33 Studies have excluded concomitant atopy and a range of possible metabolic, biochemical, and endocrine factors in disease pathogenesis. The clinical features can resemble hypertrophic lichen planus or nodular prurigo or autoimmune inflammatory blistering, or even dermatitis artefacta. Like the pretibial subtype, the initial onset of symptoms and signs may be delayed for several decades, often leading to a genetic cause for the skin lesions being erroneously discounted. Although the shins are often involved, pruritic skin lesions can occur at any site.
thought to be pathognomonic for subtypes of dominant dystrophic EB, are now recognized to occur in several forms of dystrophic EB.
Dominant dystrophic EB acral The nature of this subtype of dominant dystrophic EB is not precisely defined as the term ‘acral’ is not widely used in the published literature.31 Its inclusion in the latest classification of EB is to assist description of those cases of dominant dystrophic EB with a more localized pattern of skin involvement, usually involving the hands and feet (Fig. 4.19). However, the word acral is used in preference to localized because some oral or esophageal involvement can occur despite the relative lack of involvement of much of the skin. Trauma-induced blistering, scarring, and milia are typically present in acral skin.
Dominant dystrophic EB nails only This condition is often only first diagnosed when a family member presents with trauma-induced skin blistering, and a review of the pedigree reveals one or more generations of other individuals with nail dystrophy, but no history of blisters, scarring, milia, or mucosal erosions. Thus, in some individuals there are no clinical signs present apart from nail dystrophy, which can sometimes just be confined to the great toenails, and therefore in many families, a subtype of dystrophic EB is not suspected at all.34 This variant of dominant dystrophic EB is therefore likely to be much more common than is currently appreciated.
Dominant or recessive dystrophic EB pretibial This subtype of dystrophic EB may be inherited by either autosomal dominant (the majority) or autosomal recessive transmission with considerable overlap in appearances.32 Clinically, there are blisters, atrophy, and scarring on the shins (Fig. 4.20). Lesions are usually violaceous, sometimes mimicking lichen planus. Nails on both the hands and feet tend to be dystrophic. The shins may be itchy and the phenotype can overlap with dystrophic EB
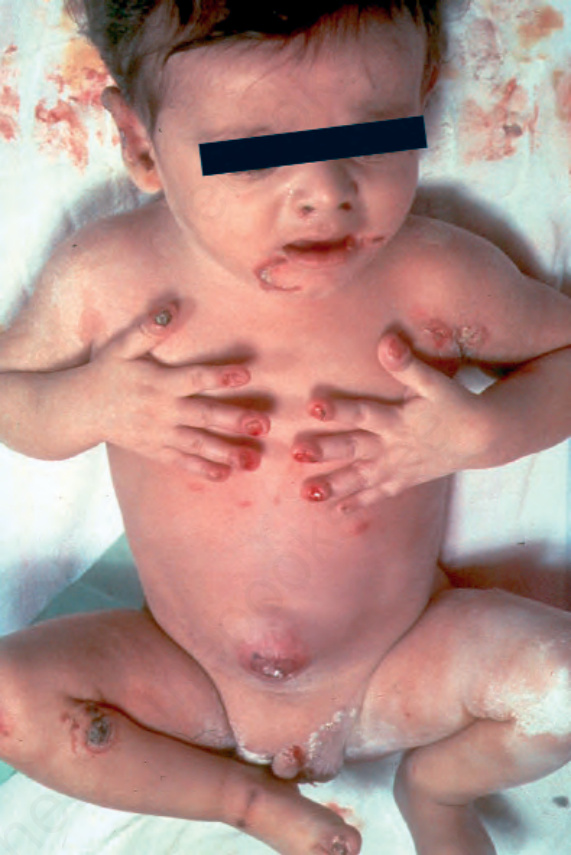
Fig. 4.14 Generalized severe junctional EB: newly born infant with blistering and nail involvement. By courtesy of J. McGrath, MD, Institute of Dermatology, London, UK.
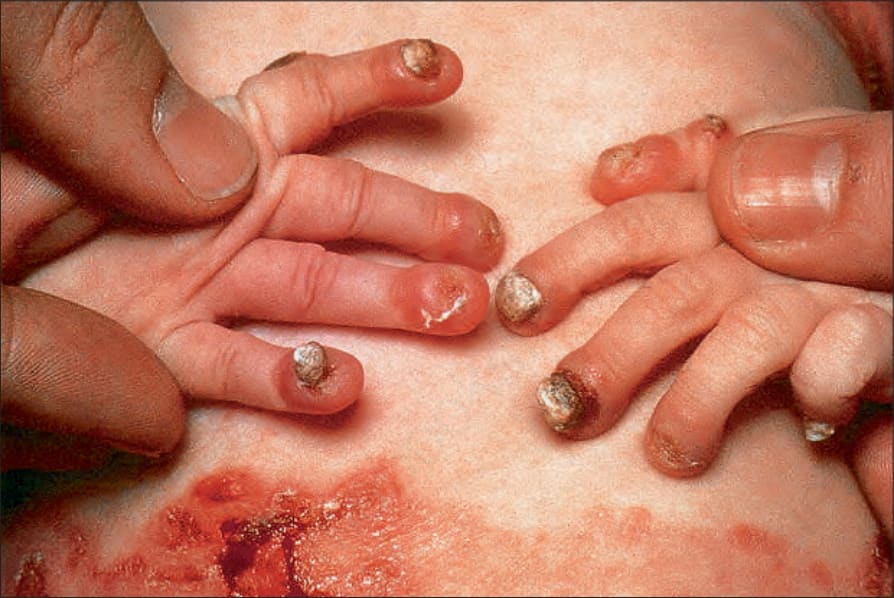
Fig. 4.15 Generalized severe junctional EB: infant showing granulation tissue at the edge of a healing blister. By courtesy of the Institute of Dermatology, London, UK.
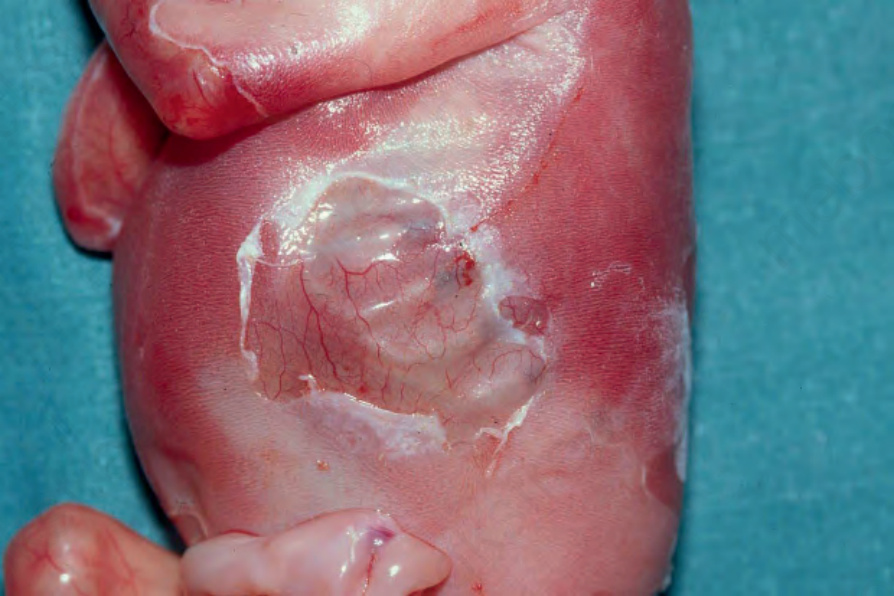
Fig. 4.16 Junctional EB with pyloric atresia: widespread blistering with deep ulceration. By courtesy of M.J. Tidman, MD, Institute of Dermatology, London, UK.
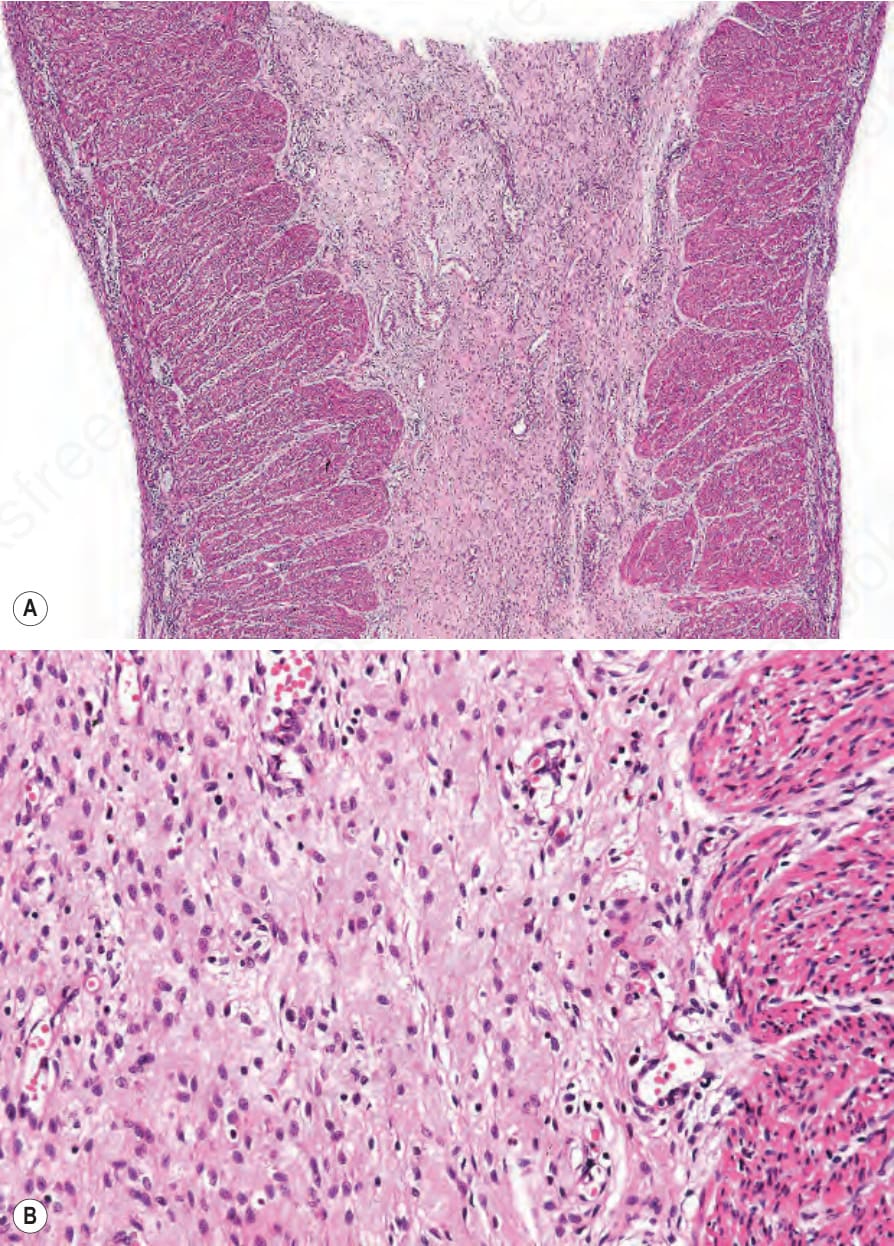
Fig. 4.17 (A, B) Junctional EB with pyloric atresia: pyloric canal is obliterated by fibrous connective tissue.
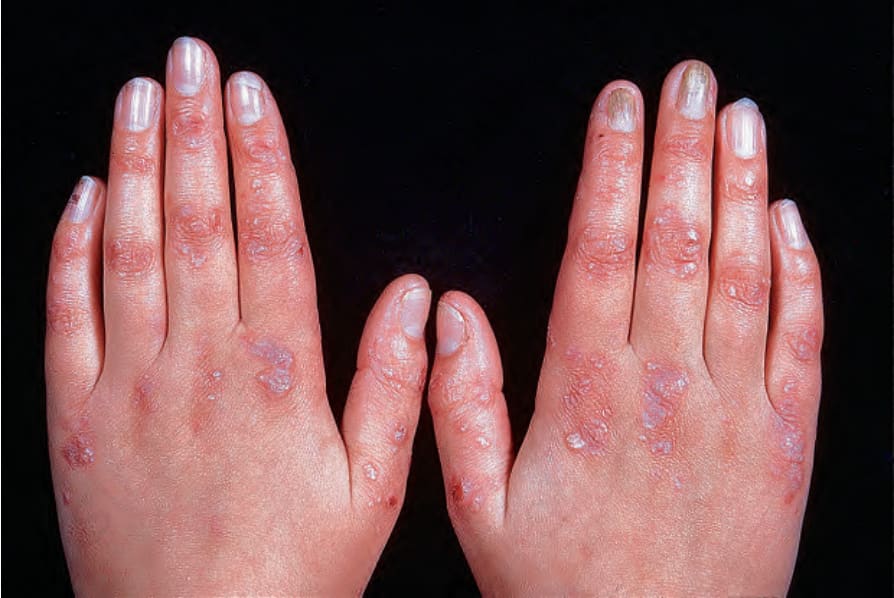
Fig. 4.18 Generalized dominant dystrophic EB: scarring, milia and nail dystrophy. By courtesy of St John’s Institute of Dermatology, London, UK.
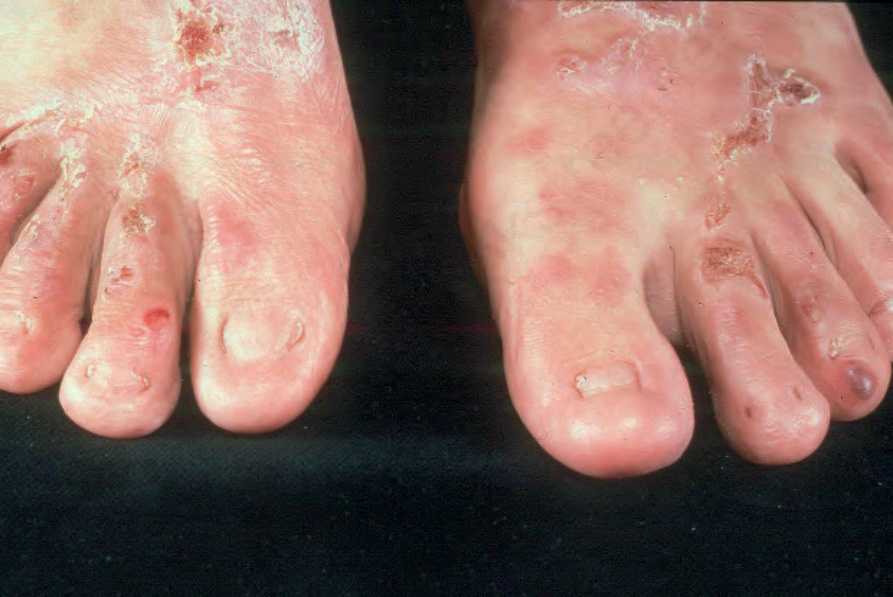
Fig. 4.19 Dominant dystrophic EB–acral: predominantly acral blisters and scarring as well as nail dystrophy. By courtesy of St John’s Institute of Dermatology, London, UK.
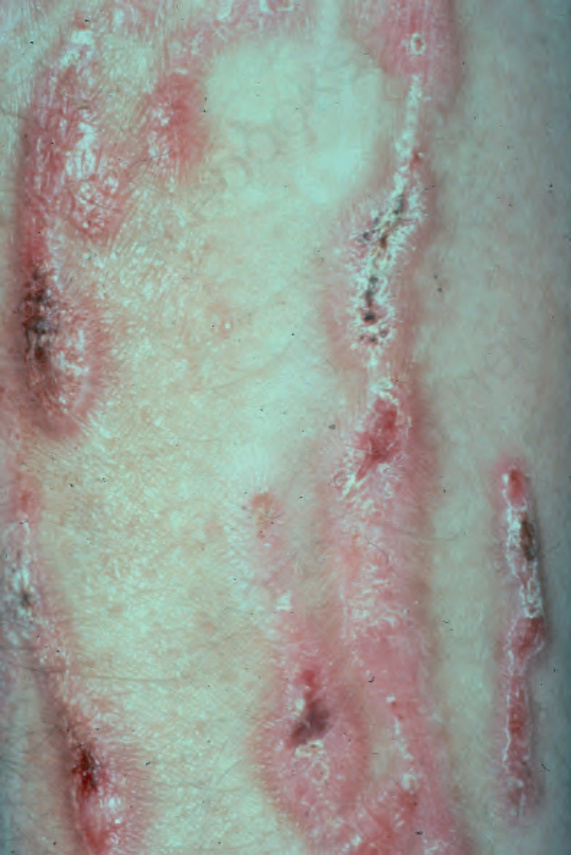
Fig. 4.20 Dominant dystrophic EB–pretibial: linear erosions with scarring localized to the front of both shins. By courtesy of St John’s Institute of Dermatology, London, UK.
Table 4.4 Clinical subtypes of dystrophic epidermolysis bullosa (EB)
Dominant or recessive dystrophic bullous disease of the newborn One curious variant of dystrophic EB is when blistering in neonates shows signs of spontaneous clinical improvement over the first few weeks or months of life (Fig. 4.21). The amelioration in phenotype is mirrored by improvement of the underlying skin pathology with increased type VII collagen at the dermal–epidermal junction. Initial skin biopsies reveal punctate intraepidermal labeling for type VII collagen and ultrastructural signs of
128 Inherited and autoimmune subepidermal blistering diseases
B A
C D
dilated perinuclear vacuoles with a granular appearance (stellate bodies). Initially, it was thought that complete correction of the type VII collagen secretion and assembly into anchoring fibrils occurred leading to the diagnostic label of ‘transient bullous dermolysis of the newborn.’35 Now, however, it is appreciated that most cases do not resolve completely and that there may be permanent stigmata of dominant or recessive dystrophic EB, albeit less severe than in early life.
occur. During childhood, repeated blistering with progressive scarring leads to fusion (‘pseudosyndactyly’) of adjacent fingers and toes (Fig. 4.22). Digits can undergo progressive contractures and gradually become encased in a cocoon-like covering of thin scar tissue, resembling a mitten.
Recessive dystrophic EB generalized severe Multiple blisters are present at birth or appear in early infancy and the skin is very fragile. The clinical presentation may include localized absence of skin especially affecting the lower legs. Blisters develop spontaneously or after the mildest trauma on any part of the skin and may be hemorrhagic.36 Milia and scarring are very common. Although any site can blister, the main areas are those subjected to repeated friction and other forms of physical trauma. These include the knees, elbows, hands, feet, back of the neck, shoulders, and over the spine. Chronic erosions and ulcers tend to become covered with a slough, often associated with heaped-up crusting and scaling, increasing the risk of secondary infection and biofilm formation. Pruritus and pain are frequent. The scalp is often involved and scarring alopecia may
Oral blistering and scarring can lead to ankyloglossia and microstomia. The gums are fragile, and gentle tooth brushing may induce epithelial disruption with bleeding. The lingual papillae are lost and the surface of the tongue becomes smooth. Although there is limited evidence for a primary abnormality of dental enamel in dystrophic EB, the teeth are at a high risk of developing caries. Blistering in the esophagus may cause acute pain and dysphagia, with difficulty in swallowing solids, with subsequent development of obstruction from strictures caused by scarring and fibrosis. Perianal blistering, erosions, and painful fissures are common in childhood. Eye complications include symblepharon, corneal erosions, and corneal opacity or scarring. Patients are often anemic and osteopaenia is not uncommon. Rarely, secondary amyloidosis can also develop in cases associated with persistent chronic inflammation and extensive scarring. A common and clinically very important complication of this form of EB is the development of squamous cell carcinomas, even in individuals as young as 6 years of age. Most carcinomas are on the limbs, often in areas of chronic, non-healing
129 Epidermolysis bullosa
of the blistering may be generalized and not indicative of the subsequent pattern.38 Traumatic corneal erosions and esophageal lesions are common. Nail dystrophy, mucous membrane involvement, and dental changes are similar to those in the generalized form of the condition. Patients are also at risk of developing squamous cell carcinoma. The reason for the predominance of flexural involvement is not clear.
Recessive dystrophic EB localized Localized forms of recessive dystrophic EB overlap with the generalized intermediate subtypes, as well as those classified as inversa, pretibial, or nails only, underscoring the spectrum of clinical features and the somewhat arbitrary goal of trying to fit all subtypes into neat, clearly defined categories. Skin fragility, scarring and milia are mostly confined to hands, feet and nails, and may be minor features.39 In some cases, onset of symptoms may be delayed for several years after birth. Mucosal involvement is rare.
ulceration (Fig. 4.23). Multiple primary tumors, with progressive loss of differentiation for each subsequent cancer is the usual course, death typically occurs within 5 years of the first malignancy.
Recessive dystrophic EB centripetalis This subtype of dystrophic EB is rare and because only one individual has been reported it is likely to be subsumed into other categories in future classifications.40 The reported individual had generalized blistering at birth but within the first year of life her skin disease activity became confined to the hands and feet. Over several decades the blistering slowly progressed proximally, in a centripetal manner, with active blistering and milia only along the active edges with atrophic scarring and nail dystrophy distally. No systemic involvement occurred.
Recessive dystrophic EB generalized intermediate There are milder subtypes of dystrophic EB that share several cutaneous and extracutaneous features with generalized severe recessive dystrophic EB, but to a much lesser degree.37 The skin and mucosae are very fragile, but lesions, including nail changes, milia, and atrophic scarring, tend to be more localized and similar to those seen in many cases of dominant dystrophic EB (Fig. 4.24). Growth retardation and anemia are usually mild. Pseudosyndactyly, esophageal involvement, and squamous cell carcinoma may also occur, but these complications are usually milder, less frequent, or have a later onset compared to generalized disease.
Recessive dystrophic EB inversa In the inversa subtype, primary areas of blistering and scarring include the groins, axillae, neck, and lumbar area, although in early life the distribution
Kindler syndrome Kindler syndrome is an autosomal recessive disorder. Initially it can resemble generalized or localized forms of dystrophic EB, although skin biopsy typically shows a variable plane of cleavage and a fragmented or duplicated lamina densa in contrast to the specific planes of tissue separation that underlie EB simplex, junctional EB, and dystrophic EB.41 Thus, Kindler syndrome is classified as a separate category of EB (Table 4.1). Clinically, the initial skin blistering lessens during childhood and instead signs of a progressive poikiloderma develop (Fig. 4.25). At this stage, the differential diagnosis includes other congenital poikilodermatous disorders, including dyskeratosis congenita and Rothmund-Thomson syndrome. Other features include gingival inflammation, ectropion, corneal erosions, chronic colitis, periodontal disease, scarring of the external urethral meatus, and an increased risk of developing cutaneous squamous cell carcinoma. The hands can also show evidence of pseudosyndactyly, similar to some cases of dystrophic EB.
130 Inherited and autoimmune subepidermal blistering diseases
Pathologic basis of EB The two key adhesion complexes implicated in the various subtypes of EB are the hemidesmosome (Fig. 4.26) and the desmosome (Fig. 4.27), and the 18 different genes implicated in the different types of EB are presented in Table 4.5.
be structural alteration in the keratin filament network resulting from some KRT5 or KRT14 mutations (Fig. 4.29). The diagnosis of EB simplex by light microscopy of paraffin-embedded sections can be challenging because early changes occur very close to the dermal–epidermal junction and may be subtle (Fig. 4.30) whereas older lesions can appear subepidermal (Fig. 4.31).
Keratins 5 and 14: KRT5, KRT14 Keratins are the most abundant structural proteins in the cytoplasm of epithelial cells.42 Pairs of keratin monomers polymerize to form a network of 10 nm-diameter intermediate filaments that maintain the shape of keratinocytes. Keratin 5 and 14 are the major keratins in basal keratinocytes. Autosomal dominant mutations in KRT5 or KRT14 underlie the most common subtype of EB, localized EB simplex, a condition affecting close to 400 000 people worldwide. EB simplex usually results in minor blistering that is typically worse in the summer months and which does not result in scarring. However, there are several other clinical variants of EB simplex that also result from KRT5 or KRT14 mutations (Table 4.2); most of these are dominant, but autosomal recessive mutations in KRT14 can occur. Blistering typically occurs within the basal keratinocyte layer (Fig. 4.28), and there may
Plakophilin-1: PKP1 Some forms of EB result from mutations in proteins found within desmosome cell–cell junctions (Fig. 4.32). Desmosomes form structural and signaling links between adjacent cells and are found in skin keratinocytes, cardiac myocytes, the meninges, and the cortex of lymph nodes. Plakophilin-1 has a restricted localization to keratinocyte desmosomes,43 and autosomal recessive loss-of-function mutations in PKP1 result in ectodermal dysplasia – skin fragility syndrome. Blistering and erosions result from a loss of keratinocyte adhesion within the desmosomal inner plaque just inside the keratinocyte (i.e., not true acantholysis); the ectodermal dysplasia partly results from altered epidermal differentiation and proliferation, but is also because plakophilin-1 has nuclear signaling roles in other tissues as well.
131 Epidermolysis bullosa
A
B
132 Inherited and autoimmune subepidermal blistering diseases
acantholysis and a poor prognosis. Autosomal recessive mutations have also been reported in individuals with Naxos disease – a combination of woolly hair, palmoplantar keratoderma, and cardiomyopathy; heterozygous carriers may also be prone to cardiac arrhythmias or heart failure. Other autosomal dominant mutations may cause cardiomyopathy.
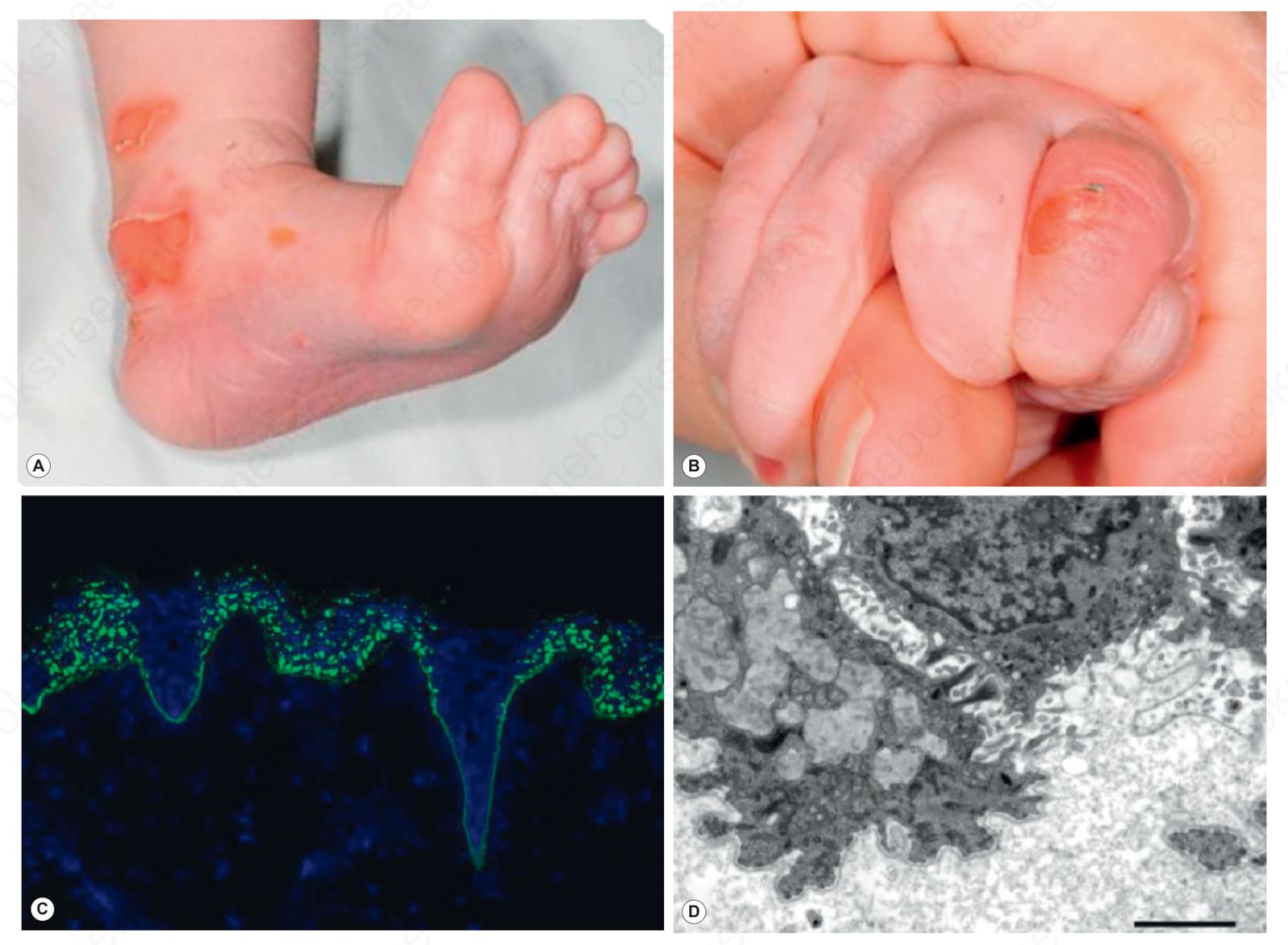
Fig. 4.21 Bullous dermolysis of the newborn: (A) blisters on the heel; (B) blisters on the fingers; (C) type VII collagen is present at the dermal–epidermal junction but there is also striking punctate staining within the epidermis; (D) ultrastructurally, in this basal keratinocyte there are numerous pale gray stellate bodies (dilated Golgi apparatus containing type VII collagen). This form of dystrophic EB usually tends to improve spontaneously during the first few months of life.
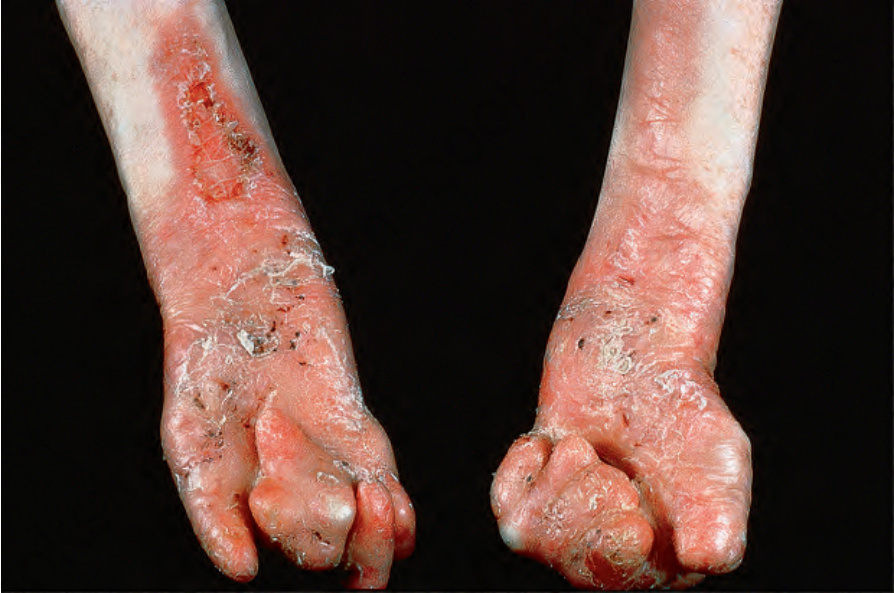
Fig. 4.22 Generalized severe recessive dystrophic EB: in addition to the gross mitten deformity, there is very severe scarring and scaling. By courtesy of St John’s Institute of Dermatology, London, UK.
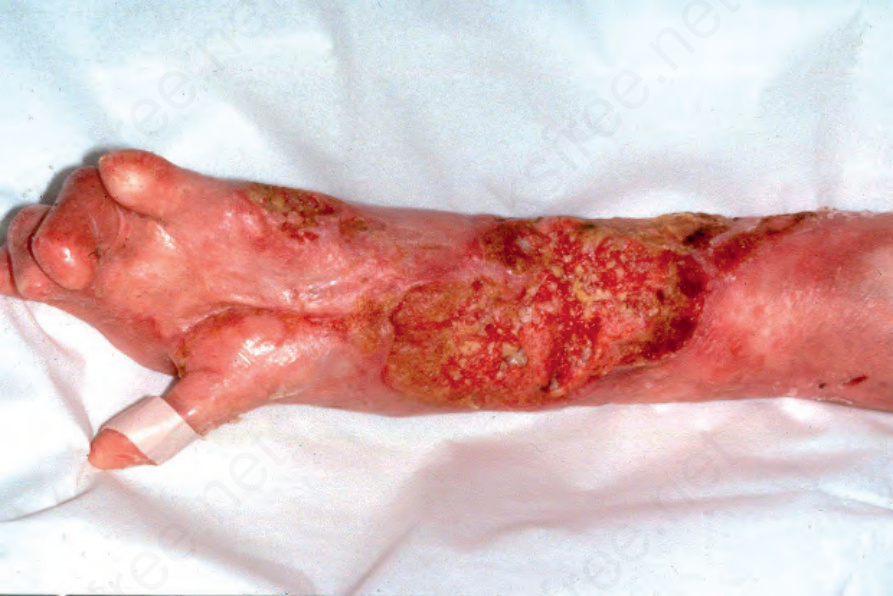
Fig. 4.23 Generalized severe recessive dystrophic EB: in this patient, numerous large keratoses are evident. Many of these progress to squamous cell carcinoma. Courtesy of R.A.J. Eady, MD, and B. Mayou, MD, St Thomas’ Hospital, London, UK.
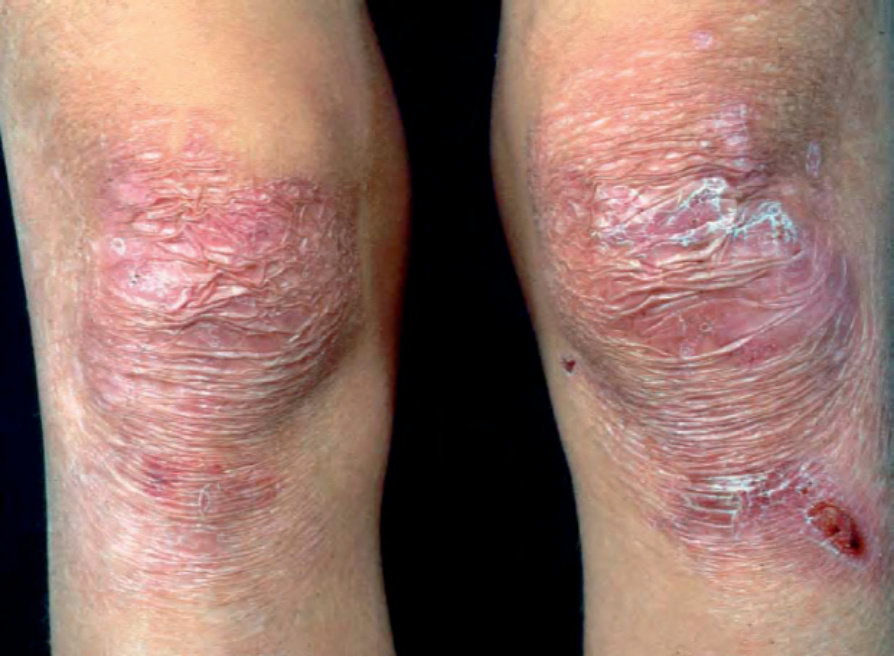
Fig. 4.24 Generalized intermediate recessive dystrophic EB: this individual has atrophic scarring and recent erosions overlying both knees.
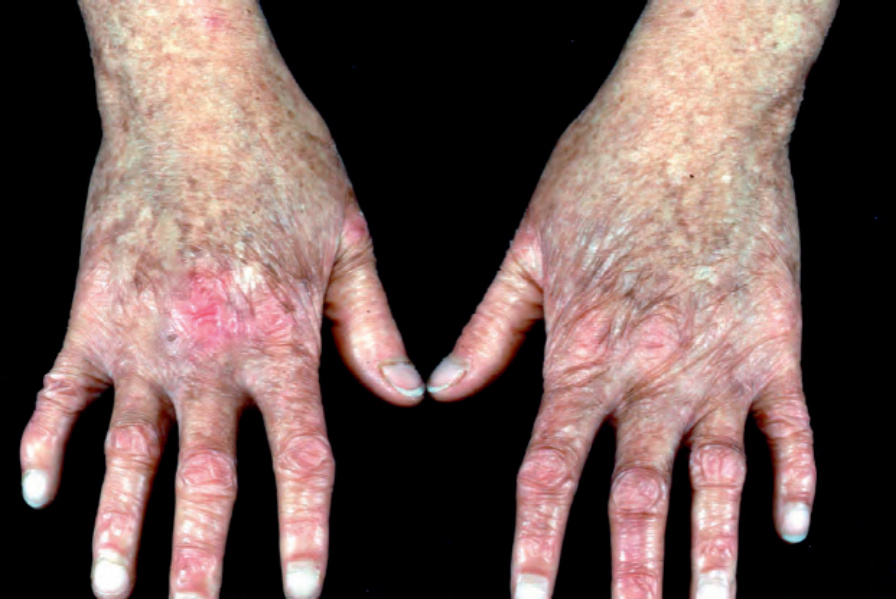
Fig. 4.25 Kindler syndrome: the hands of this 14-year-old girl show poikiloderma (hyperpigmentation, hypopigmentation, atrophy, and telangeictasias)
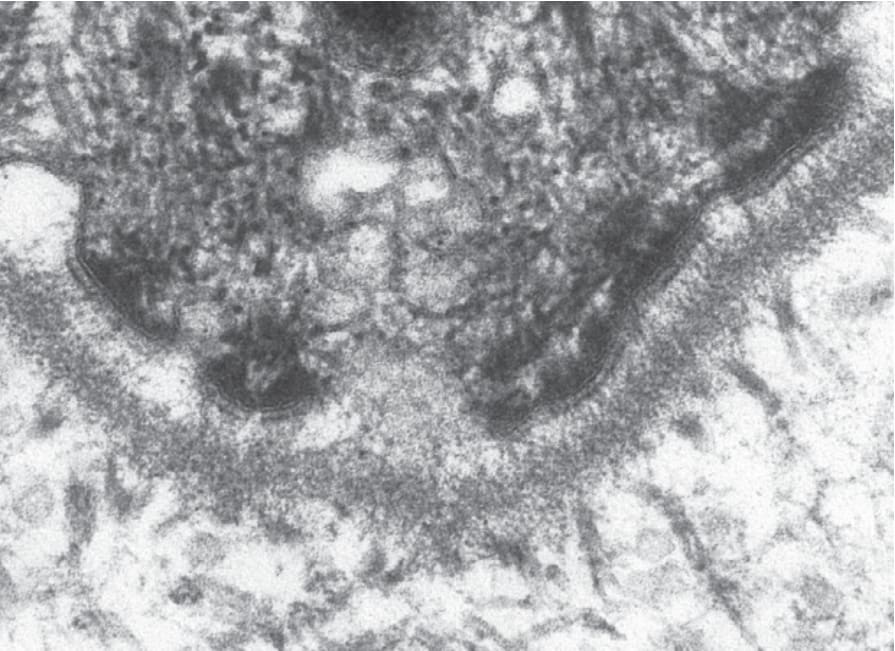
Fig. 4.26 Ultrastructural appearances of a hemidesmosome at the dermal–epidermal junction in normal human skin.
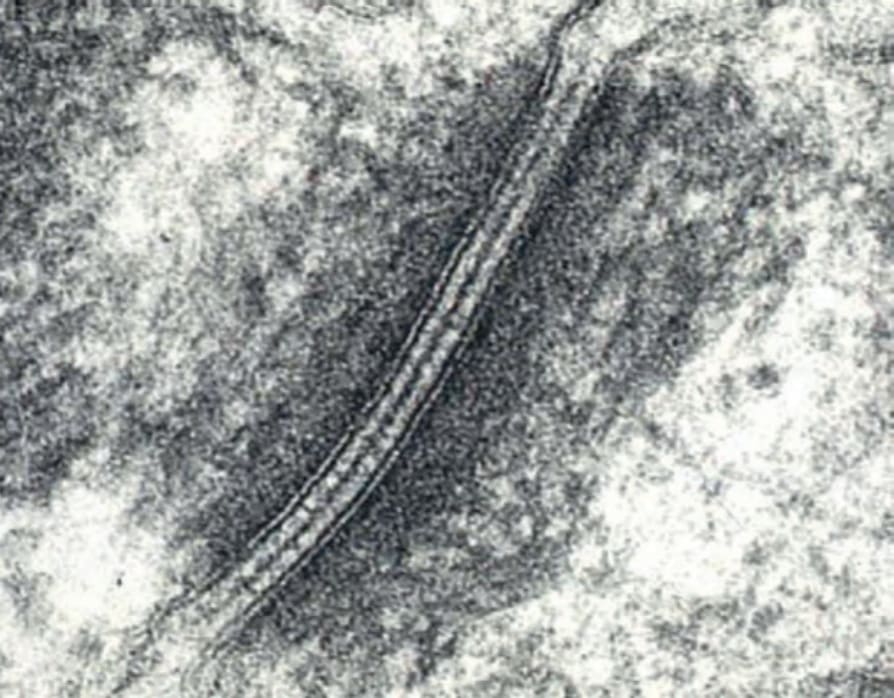
Fig. 4.27 Ultrastructural appearances of a desmosome between two keratinocytes in normal human skin.
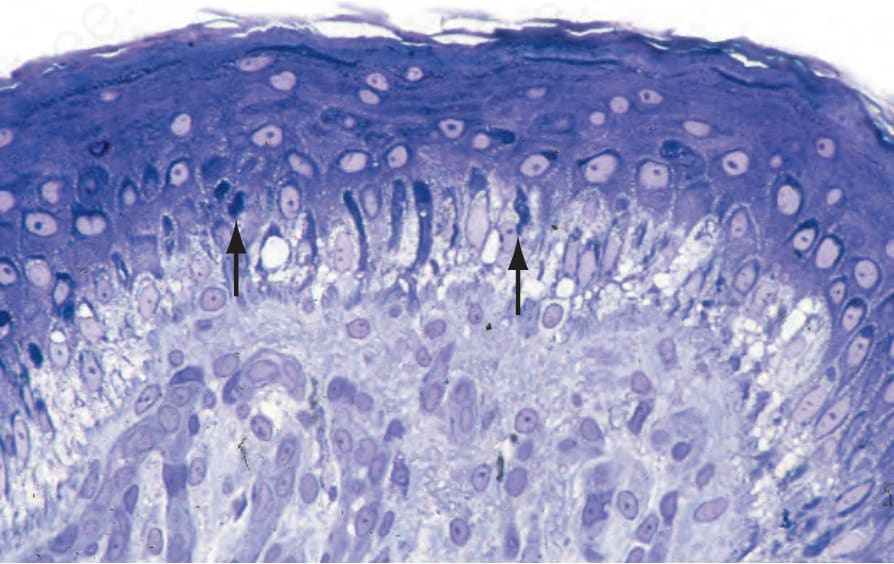
Fig. 4.28 Generalized severe EB simplex: numerous tonofilament clumps are present in the adjacent clinically normal skin (arrowed). By courtesy of J.A. McGrath, MD, St John’s Institute of Dermatology, London, UK.
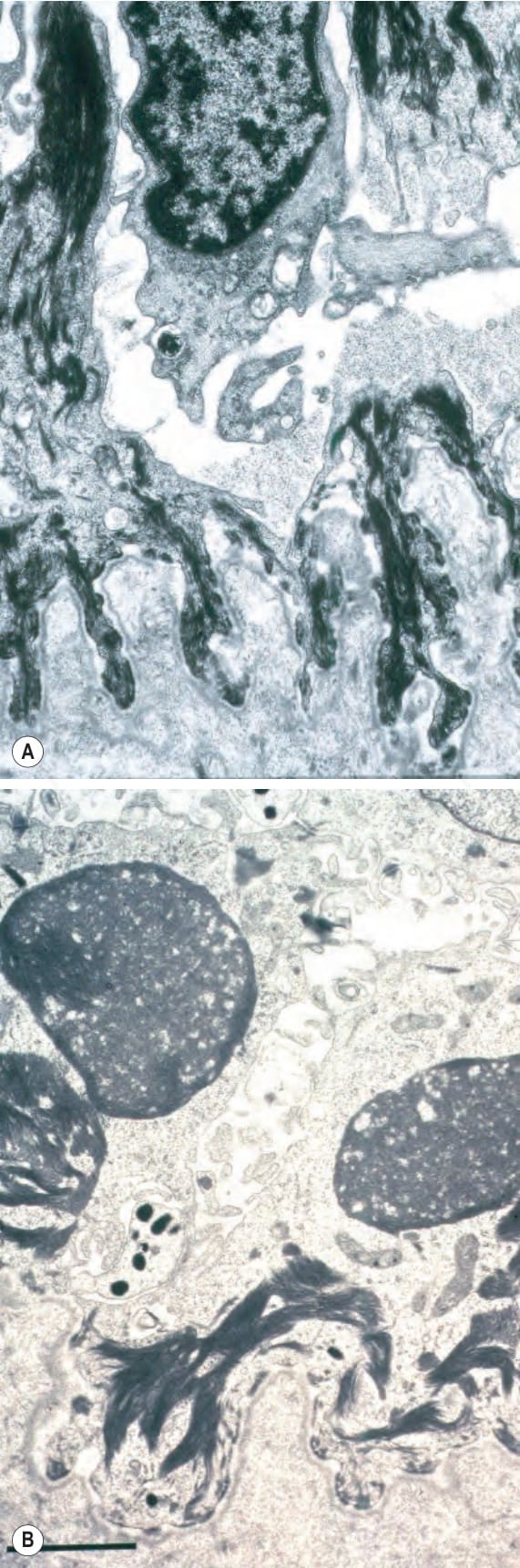
Fig. 4.29 Generalized severe EB simplex: (A) electron micrograph showing intrakeratinocyte splitting; (B) close-up view of tonofilament clumps. By courtesy of J.A. McGrath, MD, and R.A.J. Eady, MD, St John’s Institute of Dermatology, London, UK.
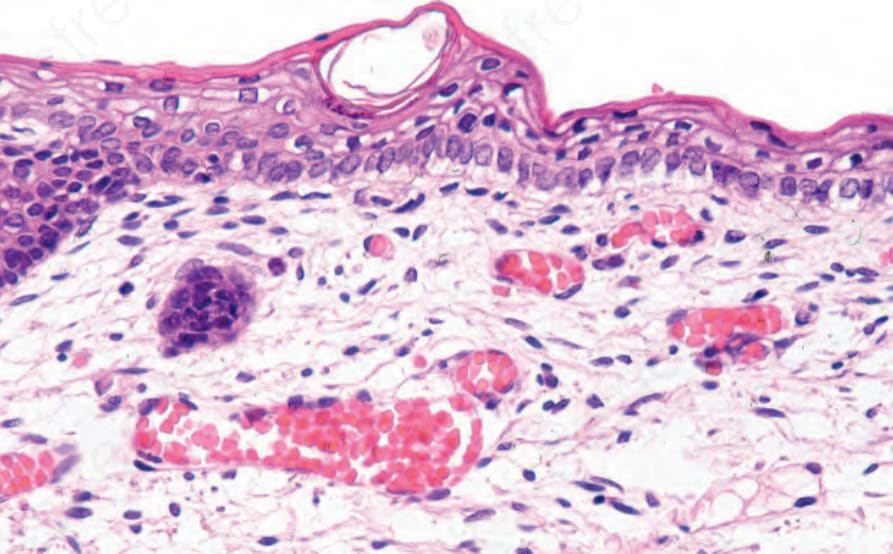
Fig. 4.30 EB simplex: the earliest histologic feature in the development of a blister is marked vacuolation of the basal keratinocytes, so-called cytolysis.
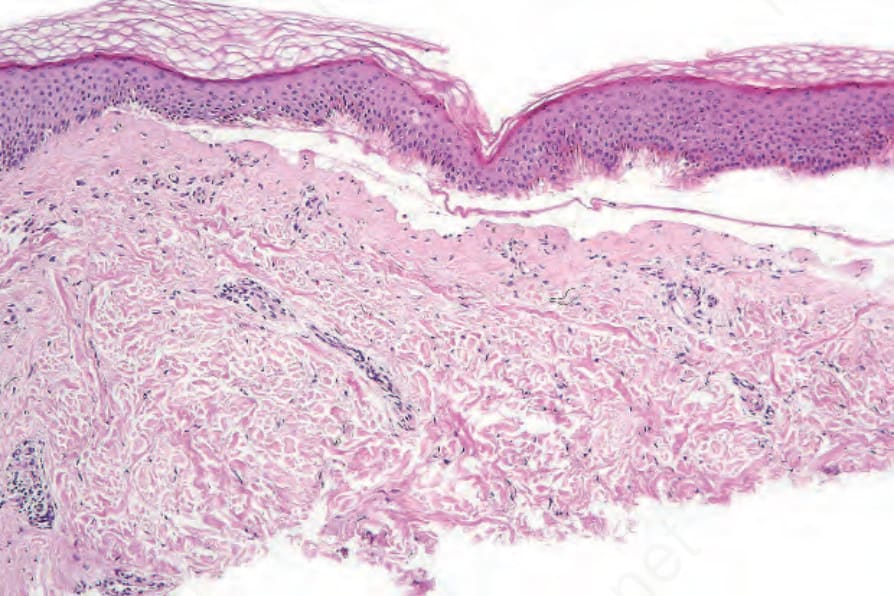
Fig. 4.31 EB simplex: old lesion; the features are those of a cell-free subepidermal blister and are not specific.
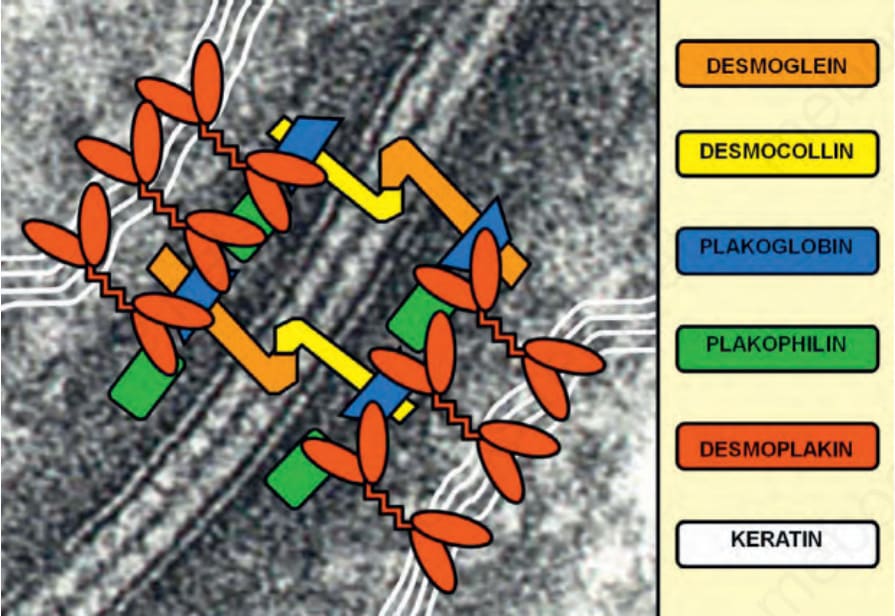
Fig. 4.32 Schematic representation of the transmembranous and intracellular components of desmosomes that provide a bridge between the keratin filament networks in adjacent keratinocytes.
Exophilin-5: EXPH5 EXPH5 encodes exophilin-5 (also known as Slac2-b), an effector protein of Rab GTPase Rab27B, which is thought to have an important role in intracellular vesicle trafficking along actin and tubulin networks, as well as in the transfer of vesicles to cell membranes.46 Loss-of-function mutations in EXPH5 lead to keratin filament clumping, cytolysis, acantholysis and increased cytoplasmic (perinuclear) vesicles. This form of inherited skin fragility has been classified as an autosomal recessive form of EB simplex.
Transglutaminase 5: TGM5 Autosomal recessive mutations in TGM5 underlie acral peeling skin syndrome. Transglutaminase 5 is one of eight different transglutaminase enzymes expressed in the skin and has a distinct role in the formation of the cornified cell envelope.47 The level of blister formation occurs above the granular layer, just below the stratum corneum. However, given the increased thickness of the stratum corneum in the palms and soles relative to other body sites, the clinical appearances often resemble the most common form of localized EB simplex and indeed may cause some clinical confusion, although both are included within the latest classification of EB (Table 4.2).
Plectin: PLEC Plectin is an epidermal plakin protein, also found within the z-lines of striated muscle.48 Autosomal recessive mutations in PLEC cause EB simplex associated with muscular dystrophy, which manifests as relatively minor skin blistering and progressive muscle weakness. Autosomal recessive mutations in PLEC can also cause skin blistering with pyloric atresia, or, occasionally, both manifestations, or sometimes just skin blistering. Autosomal dominant mutations in plectin may also occur in other forms of localized EB simplex (up to 10% of all cases), including one of the few eponymous variants in the current classification of EB, the Ogna subtype.
Desmoplakin: DSP
Desmoplakin is the major intracellular component of the desmosomal plaque.44 Autosomal recessive mutations can result in devastating mucocutaneous skin fragility, notably in a severe acantholytic form of EB. Officially this condition is classified as a suprabasal form of EB simplex, although desmoplakin has a pan-epidermal expression and all layers of the epidermis show acantholysis (Fig. 4.33) with ultrastructural cleavage through the inner plaque of desmosomes (Fig. 4.34). Loss of desmoplakin expression in these cases leads to early death because of the profound skin loss and potential involvement of other organs, notably the heart. Other autosomal recessive mutations may result in woolly hair and keratoderma but no skin fragility, and autosomal dominant mutations in DSP can also give rise to striate palmoplantar keratoderma or arrhythmogenic cardiomyopathy.
Dystonin epidermal isoform (BP230): DST-e Autosomal recessive mutations in the epidermal isoform of dystonin, also known as the 230 kDa bullous pemphigoid antigen (BP230),49 result in a relatively mild form of EB simplex. Ultrastructurally, there is a complete absence of the hemidesmosomal inner plaques – the sites at which keratin intermediate filaments anchor to the hemidesmosomes. Although dystonin isoforms have a wide tissue distribution, neurologic or cardiac involvement does not appear to be a clinical feature – predominantly acral skin blistering is the main abnormality.
α6β4 integrin: ITGA6, ITGB4 The α6β4 integrin is a cell adhesion dimer involved in hemidesmosome assembly and in epithelial–mesenchymal signaling.50 Mutations in ITGA6 or ITGB4 (that encode the α6 and β4 integrin subunits, respectively) result in autosomal recessive junctional EB associated with pyloric atresia. The clinical severity of both the skin fragility and degree of gastric obstruction can vary, but surgical correction of the pylorus is usually required. More severe forms of the disease result from loss-of-function mutations on both alleles of ITGA6 or ITGB4 although missense mutations in certain critical cysteine residues may also have devastating clinical consequences. Other missense mutations can result in different forms of generalized intermediate junctional EB.
Plakoglobin: JUP Plakoglobin is an intracellular armadillo protein component of the desmosome.45 Autosomal recessive mutations can cause an acantholytic form of EB simplex. These cases are similar to some DSP mutations – with the condition classified as a suprabasal form of EB simplex, but with pan-epidermal
α3 integrin subunit: ITGA3 The α3 integrin subunit is a component of focal contacts at the dermal– epidermal junction, where it may dimerize with β1 integrin, and contribute to epithelial–mesenchymal signaling.51 Autosomal recessive loss-of-function mutations in ITGA3 have been reported in individuals with pulmonary inflammation and congenital nephrotic syndrome, reflecting the important
role of α3 integrin in lung and kidney biology. Of note, skin blistering was relatively trivial and not always present. Prognosis is poor because of lung/ kidney disease.
Kindlin-1: KIND1/FERMT1 Kindlin-1, also known as fermitin family homolog-1, is a component of focal contacts at the dermal–epidermal junction, and has a role in anchorage of the actin cytoskeleton and formation of a signaling platform via β1 integrin.52 Autosomal recessive mutations in KIND1/FERMT1 result in Kindler syndrome, a blistering genodermatosis that may resemble dystrophic EB in early life but with increasing age, the blistering often diminishes and new features of photosensitivity and poikiloderma (a combination of hyperpigmentation, hypopigmentation, telangiectases and skin atrophy), develop, mostly in sun-exposed areas. Individuals with Kindler syndrome also have an increased risk of squamous cell carcinoma.
133 Epidermolysis bullosa
Type VII collagen: COL7A1
Type VII collagen is the major component of anchoring fibrils, adhesion complexes inserting into the dermal side of the lamina densa that are traversed by dermal collagen fibers to secure adhesion between the epidermis and dermis (Fig. 4.36).55 Mutations in COL7A1 underlie both autosomal dominant and autosomal recessive forms of dystrophic EB. Typically, loss-of-function mutations on both alleles of COL7A1 underlie the generalized severe forms of recessive dystrophic EB in which anchoring fibrils are structurally defective or completely absent (Fig. 4.37). Poor wound healing results in chronic wounds, mutilating scar formation and an increased incidence of squamous cell carcinoma, often with multiple primary tumors over a few year period (Figs 4.38 and 4.39). Initial tumors may be well-differentiated or verrucous and difficult to distinguish from pseudoepitheliomatous hyperplasia, although successive tumors become progressively less well differentiated. However, there is a spectrum of clinical severity with less disruptive mutations giving rise to less severe intermediate or localized phenotypes (Table 4.4). Dominant dystrophic EB is usually clinically milder than recessive disease and most cases result from heterozygous missense mutations within the type VII collagen triple helix.
Type XVII collagen: COL17A1 Type XVII collagen, also known as the 180 kDa bullous pemphigoid antigen, is a transmembranous protein located within the hemidesmosome and lamina lucida.53 It is the antigenic target in the autoimmune blistering disease bullous pemphigoid, but loss-of-function mutations on both alleles result in generalized intermediate junctional EB (previously known as non-Herlitz or generalized atrophic benign EB). Some dominant missense mutations in type XVII collagen may result in defective dental enamel and occasionally skin fragility, but most pathogenic mutations in COL17A1 are autosomal recessive.
Laminin-332: LAMA3, LAMB3, LAMC2 Laminin-332, previously known as laminin-5, is a heterotrimeric protein consisting of α3, β3, and γ2 laminin polypeptide chains located within the lamina lucida/lamina densa of the epidermal basement membrane.54 Autosomal recessive mutations give rise to generalized severe (previously known as Herlitz junctional EB), generalized intermediate, or more localized forms of junctional EB (Table 4.3). The plane of tissue cleavage is through the lamina lucida at the dermal–epidermal junction (Fig. 4.35). Generalized severe disease is associated with widespread mucocutaneous fragility and a poor prognosis. Clinically less severe forms of junctional EB are usually associated with mutations that allow for some residual functional laminin-332 protein. Mutations in the LAMA3A isoform of the LAMA3 gene are associated with laryngo-onycho-cutaneous syndrome, in which excessive granulation tissue can lead to laryngeal obstruction and blindness.
134 Inherited and autoimmune subepidermal blistering diseases
Fig. 4.33 Desmosomal EB simplex: pan-epidermal cell-cell detachment within the epidermis, here resulting from autosomal recessive mutations in desmoplakin.
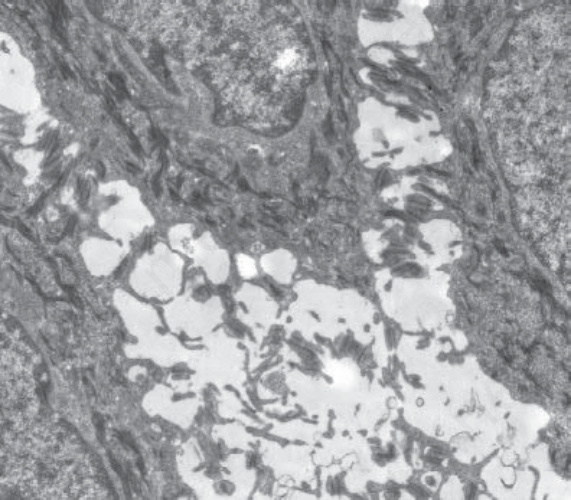
Fig. 4.34 Desmosomal EB simplex: the ultrastructural plane of cleavage leading to cell separation occurs through the intracellular desmosomal plaque consistent with the localization of the mutant desmoplakin in this case.
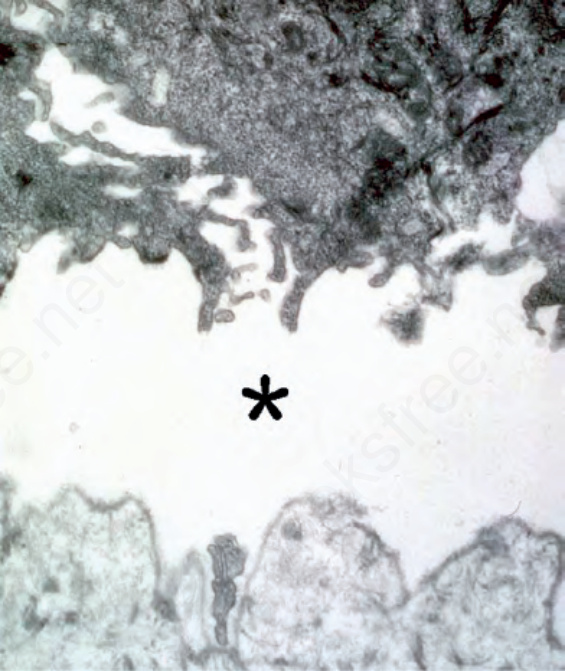
Fig. 4.35 Junctional EB: the level of blister formation at the dermal–epidermal junction is through the lamina lucida (asterisk).
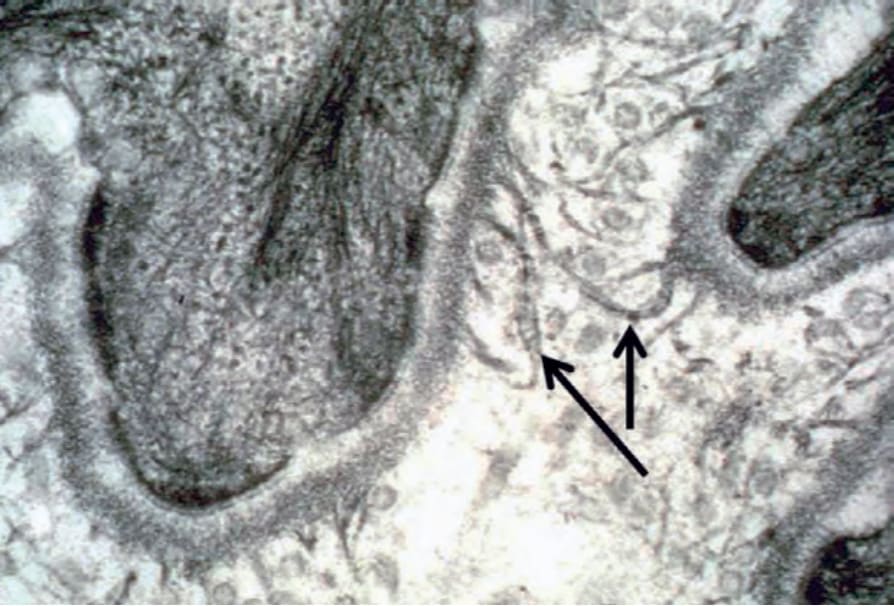
Fig. 4.36 Anchoring fibrils in normal skin: fibrillar structures with a fan-shaped appearance, central cross-banding and insertion into the lamina densa represent the ultrastructural hallmarks of anchoring fibrils in the superficial dermis (arrows).
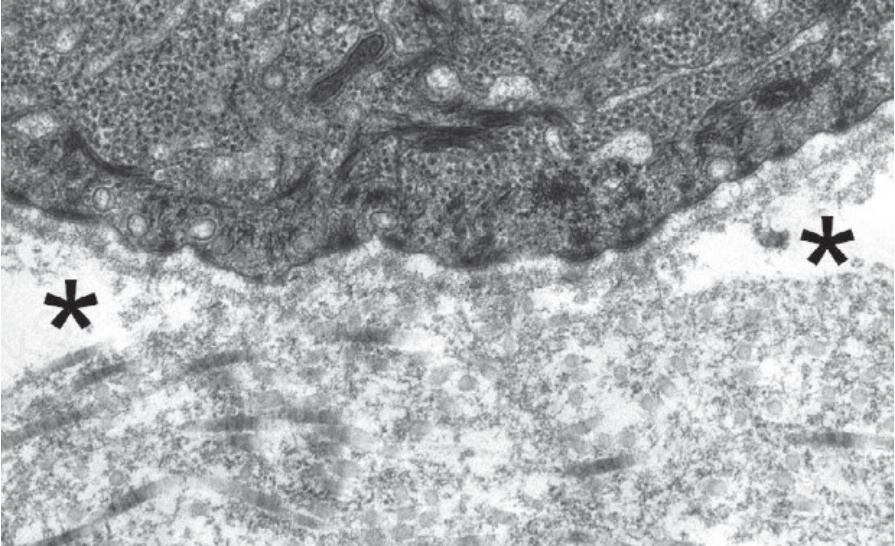
Fig. 4.37 Generalized severe recessive dystrophic EB: complete absence of anchoring fibrils below the lamina densa with onset of sub-lamina densa blistering (asterisks).
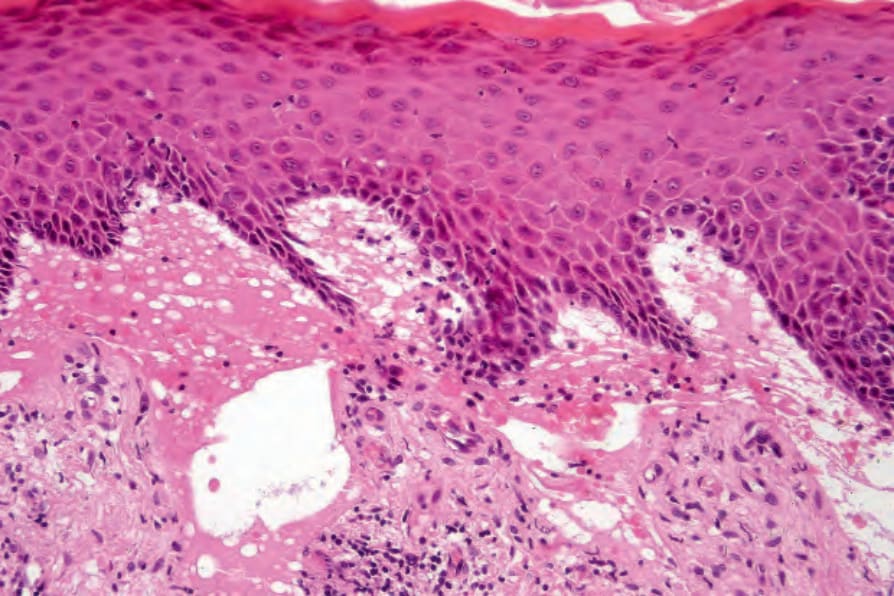
Fig. 4.38 Generalized severe recessive dystrophic EB: in addition to obvious subepidermal blistering there is dermal scarring and chronic inflammation.