Richner-Hanhart 症候群 (Richner-Hanhart Syndrome)
臨床特徵 (Clinical Features)
- Richner-Hanhart 症候群(眼皮膚型酪胺酸血症 [oculocutaneous tyrosinemia]、tyrosine transaminase deficiency、第 II 型酪胺酸血症 [tyrosinemia type II])為一種眼皮膚症候群,特徵為生命最初數月內發生的疱疹樣角膜潰瘍 (herpetiform corneal ulcers)。其後出現指(趾)、掌、蹠之疼痛性界限分明的角化過度 (circumscribed hyperkeratoses),典型上致使孩童以足趾行走 (walk on the toes)。可能發生水疱與多汗 (hyperhidrosis)。偶有報告於肘、膝甚至舌部出現角化性斑塊。其他症狀包括嚴重的智能與身體發育遲滯 (mental and somatic retardation)。
致病機轉與組織學特徵 (Pathogenesis & Histologic Features)
- 第 II 型酪胺酸血症 (Tyrosinemia type II) 係由體染色體隱性遺傳之肝臟 tyrosine aminotransferase 缺乏所致,原因為 tyrosine aminotransferase 基因 TAT 之點突變 (point mutations)。裂隙燈檢查 (slit-lamp examination) 可見酪胺酸結晶沉積 (tyrosine crystal deposition),且血清與尿中酪胺酸濃度升高。限制酪胺酸與苯丙胺酸 (tyrosine and phenylalanine restricted diet) 之飲食可清除角膜炎 (keratitis) 與皮膚病灶,並可能延緩或預防認知障礙。
- 組織學上,表皮呈棘層肥厚 (acanthotic),顆粒層增厚,角質細胞含有嗜伊紅性球狀內含物 (eosinophilic globular inclusions),與 PC 所見者相似。
- 電子顯微鏡顯示成簇的張力絲 (clumped tonofilaments) 伴附著之球狀角質透明顆粒 (globoid keratohyalin granules),提示因細胞內酪胺酸過量而增強微絲聚集 (microfilament aggregation),並伴有針狀酪胺酸結晶內含物 (needle-shaped tyrosine crystalline inclusions)。
掌蹠點狀角化症 Buschke-Fischer-Brauer 型 (Keratosis Punctata Palmoplantaris Type Buschke-Fischer-Brauer)
臨床特徵 (Clinical Features)
- 點狀掌蹠角皮症 (punctate palmoplantar keratoderma) 之特徵為掌、蹠上的小型圓形丘疹狀角化 (rounded papular keratoses)。有三個基因上互不相關的病種,分類為掌蹠角皮症點狀型第 1–3 型 (palmoplantar keratoderma punctata type 1–3)。
- 掌蹠點狀角化症 Buschke-Fischer-Brauer 型(keratosis punctata palmaris et plantaris、keratoderma hereditarium dissipatum palmare et plantare、Davis-Colley disease)現分類為掌蹠角皮症點狀型第 1 型 (palmoplantar keratoderma punctata type 1, PPKP1)。PPKP1 以體染色體顯性方式遺傳。病人於整個掌蹠面發生眾多微小的角化性丘疹。中央角化核 (central keratotic core) 呈半透明,可變為不透明或疣狀 (verrucous),移除後留下凹陷的小坑 (depressed pit)(圖 3-123)。在足蹠的受壓點,丘疹聚集成大型的角化性斑塊(圖 3-124)。發病多在第一、二十年(即十歲與二十歲年齡層),或可延遲至第六十年(即五十多歲),具有相當大的家族間與家族內變異性。有時病灶與過度的手工勞動相關。偶可出現疼痛、壓痛或灼熱感。PPPK1 是否真正與惡性腫瘤相關尚不清楚。
致病機轉與組織學特徵 (Pathogenesis & Histologic Features)
- PPPK1 已被定位至兩個染色體區域 15q22 及 8q24.13–8q24.21。第一個基因座 (locus) 含有 AAGAB 基因,其編碼 alpha- 與 gamma-adaptin-binding protein p34,參與網格蛋白被覆囊泡 (clathrin-coated vesicle) 之運輸。p34 缺乏導致表皮生長因子 (epidermal growth factor) 訊息傳遞增加,進而可能驅動角質細胞增生。第二個位於 8q 的基因座,於一中國家族中被發現與 COL14A1 基因之一個錯義突變相關。
- 組織病理學上,出現伴正角化過度 (orthohyperkeratosis) 的表皮凹陷 (epidermal depression)(圖 3-125)。其下方可見顆粒層減少 (hypogranulosis) 伴局部角化不全 (parakeratosis),以及拉長且彎曲的表皮突 (rete ridges),常被誤診為尋常疣 (verruca vulgaris)(圖 3-126)。其他 HPV 感染的細胞學特徵則付之闕如。電子顯微鏡顯示大量小型囊泡與擴張的高基氏體 (Golgi apparatus),凸顯囊泡運輸 (vesicle trafficking) 的失調。
鑑別診斷 (Differential Diagnosis)
- 後天型點狀掌蹠角皮症 (acquired forms of punctate palmoplantar keratoderma) 可能與內臟惡性腫瘤相關,包括結腸、腎、乳房及胰臟之癌,以及 Hodgkin lymphoma。曾有報告砷 (arsenic) 或戴奧辛 (dioxin) 暴露之併發症會出現類點狀角皮症 (punctate keratoderma-like) 之病灶累及掌與蹠。掌紋點狀角化症 (Keratosis punctata of the palmar creases) 發生於黑人成人,且僅侷限於指(趾)與蹠之皺褶(見下文)。可與 PPK1 混淆者尚有基底細胞癌母斑症候群 (nevoid basal cell carcinoma syndrome)、Darier disease 或其他狀況所見之掌蹠小坑 (palmoplantar pits)。
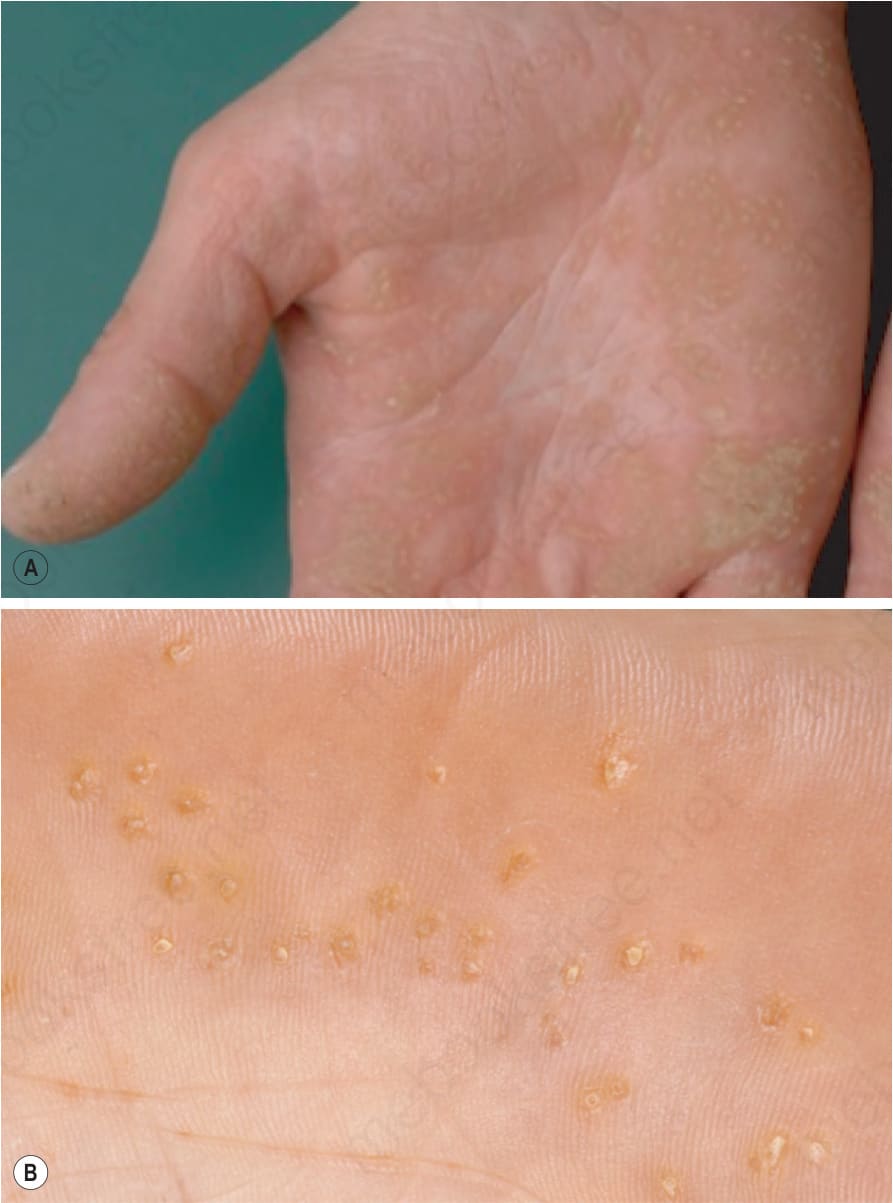
圖 3-123:點狀掌蹠角皮症 (punctate palmoplantar keratoderma):整個手掌散布微小的角化性丘疹。中央角化核呈 (A) 半透明而不透明,或 (B) 疣狀 (verrucous)。
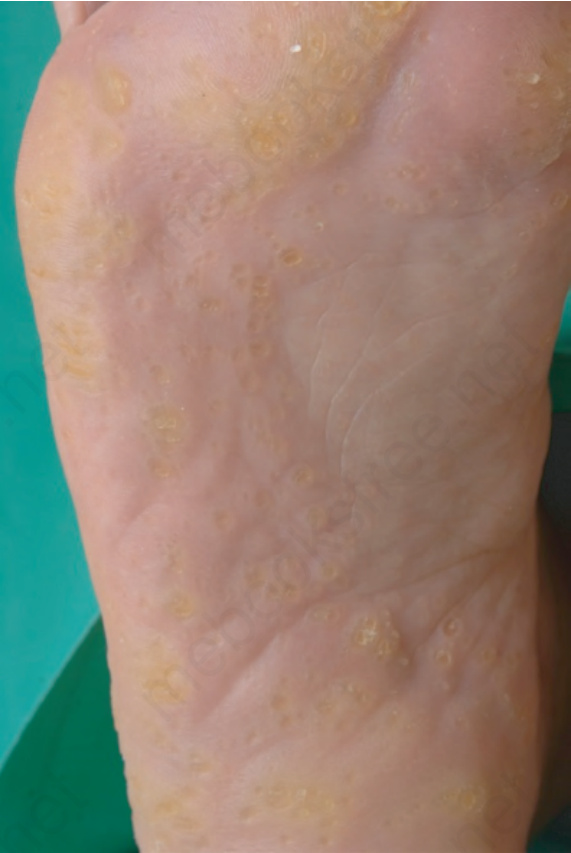
圖 3-124:點狀掌蹠角皮症 (punctate palmoplantar keratoderma):受壓部位上離散的黃色角化性丘疹融合成較大的斑塊。
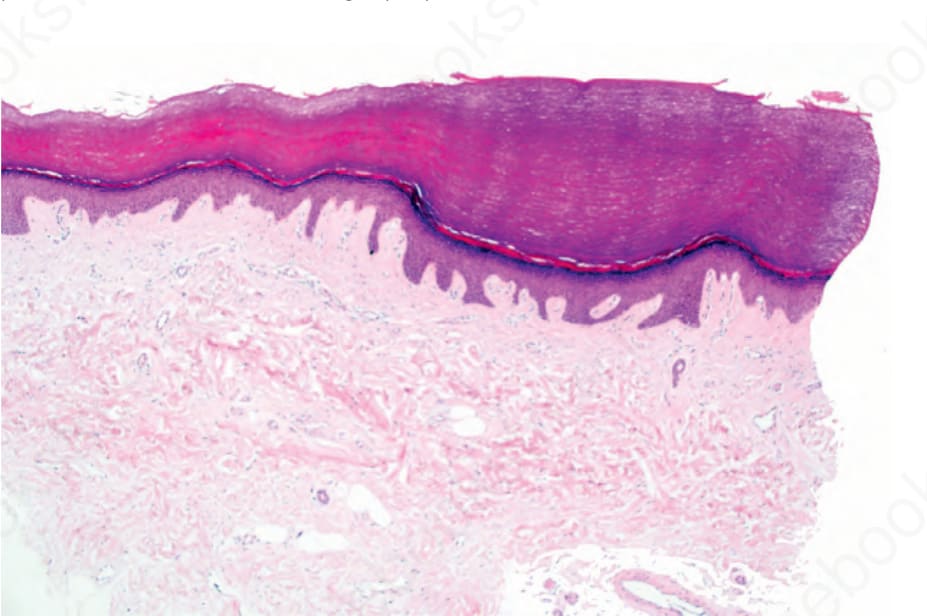
圖 3-125:點狀掌蹠角皮症 (punctate palmoplantar keratoderma):表皮凹陷之上覆蓋大量正角化過度 (orthohyperkeratosis)。
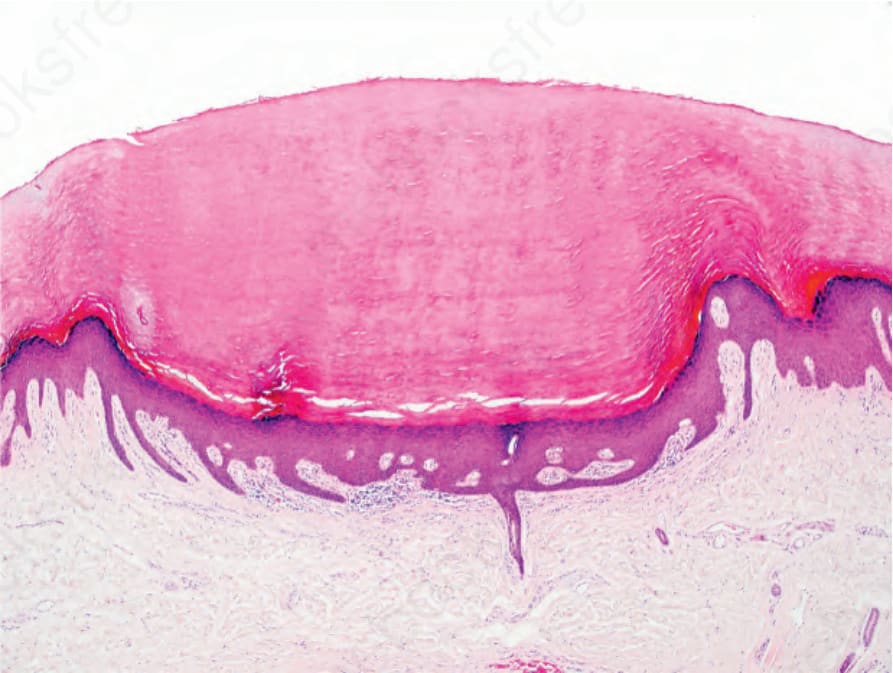
圖 3-126:點狀掌蹠角皮症 (punctate palmoplantar keratoderma):可見拉長且彎曲的表皮突 (rete ridges),常被誤診為尋常疣 (verruca vulgaris)。