Richner-Hanhart syndrome
Richner-Hanhart syndrome
Clinical features Richner-Hanhart syndrome (oculocutaneous tyrosinemia, tyrosine transaminase deficiency, tyrosinemia type II) is an oculocutaneous syndrome characterized by herpetiform corneal ulcers that develop during the first months of life.1 Later painful circumscribed hyperkeratoses of digits, palms, and soles evolve, typically making the child walk on the toes. Blisters and hyperhidrosis may occur. Keratotic plaques have been described sporadically on the elbows, knees, and even the tongue. Other symptoms include severe mental and somatic retardation.1,2
A
Pathogenesis and histologic features Tyrosinemia type II is caused by autosomal recessively inherited deficiency of hepatic tyrosine aminotransferase due to point mutations in the tyrosine aminotransferase gene TAT.3,4 Tyrosine crystal deposition is seen on slit-lamp examination, and serum and urinary tyrosine levels are elevated. A tyrosine and phenylalanine restricted diet clears the keratitis and the skin lesions, and may delay or prevent cognitive impairment.5
B
Fig. 3. 122 Pachyonychia congenita: (A) scanning view of the oral mucosa showing massive acanthosis with large blunt rete ridges; (B) high-power view showing focal parakeratosis and vacuolization of superficial keratinocytes. A single dyskeratotic cell is evident (arrowed).
Histologically the epidermis is acanthotic, the granular layer is thickened, and the keratinocytes contain eosinophilic globular inclusions similar to those seen in PC.6
Electron microscopy reveals clumped tonofilaments with adherent globoid keratohyalin granules suggesting enhanced microfilament aggregation due to an excessive amount of intracellular tyrosine with needle-shaped tyrosine crystalline inclusions.6
Keratosis punctata palmoplantaris type Buschke-Fischer-Brauer
Clinical features Punctate palmoplantar keratoderma is characterized by small rounded papular keratoses on the palms and soles. There are three genetically unrelated entities classified as palmoplantar keratoderma punctata type 1–3.
Keratosis punctata palmoplantaris type Buschke-Fischer-Brauer (keratosis punctata palmaris et plantaris, keratoderma hereditarium dissipatum palmare et plantare, Davis-Colley disease) is now classified as palmoplantar keratoderma punctata type 1 (PPKP1).1–4 PPKP1 is inherited in an autosomal dominant mode. Patients develop numerous tiny keratotic papules over the entire palmoplantar surfaces. The central keratotic core is translucent, may become opaque or verrucous and leaves a depressed pit after removal (Fig. 3.123).5 Over pressure points of the soles the papules aggregate into large keratotic plaques (Fig. 3.124). Disease onset is during the first and second decade, or may be delayed up to the sixth decade of life, with large inter- and intrafamilial variability.4 Sometimes, lesions are associated with excessive manual labor.2 Occasionally pain, tenderness or burning may occur.2,5 It is not clear whether PPPK1 is truly associated with malignancies.
103 Palmoplantar keratoderma
A
B
Pathogenesis and histologic features PPPK1 has been mapped to two chromosomal regions 15q22 and 8q24.13–8q24.21. The first locus harbors the AAGAB gene that encodes the alpha- and gamma-adaptin-binding protein p34 which is involved in the clathrin-coated vesicle trafficking.6–8 Deficiency of p34 results in increased epidermal growth factor signaling, which in turn may drive keratinocyte proliferation.9 The second locus on 8q was found to be associated with one missense mutation in the gene COL14A1 in a Chinese family.10
Histopathologically, an epidermal depression with orthohyperkeratosis appears (Fig. 3.125). Underlying hypogranulosis with focal parakeratosis and elongated and curved rete ridges may be seen and are often misdiagnosed as verruca vulgaris (Fig. 3.126). Other cytologic features of HPV infection are lacking. Electron microscopy reveals a high number of small vesicles and dilated Golgi apparatus highlighting the dysregulation of vesicle trafficking.9
Differential diagnosis Acquired forms of punctate palmoplantar keratoderma may be associated with internal malignancies, including carcinomas of the colon, kidney, breast, and pancreas, and Hodgkin lymphoma.11,12 Punctate keratoderma-like lesions affecting the palms and the soles have been described as a complication of arsenic or dioxin exposure.13 Keratosis punctata of the palmar creases occurs in black adults and is restricted to the creases of the digits and soles (see below).14 Palmoplantar pits as seen in nevoid basal cell
carcinoma syndrome, Darier disease or other conditions can be confused with PPK1.
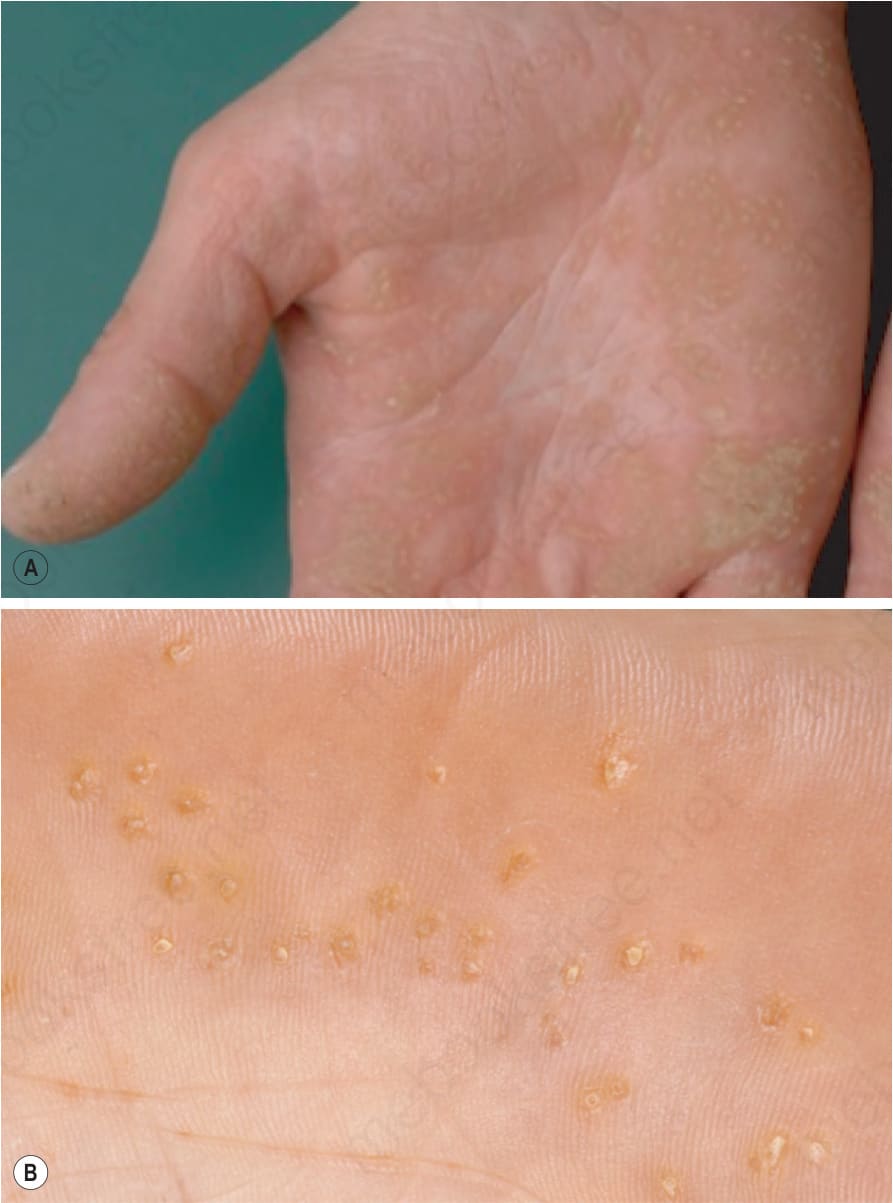
Fig. 3.123 Punctate palmoplantar keratoderma: there are tiny keratotic papules over the entire palms. The central keratotic core is (A) translucent and opaque or (B) verrucous.
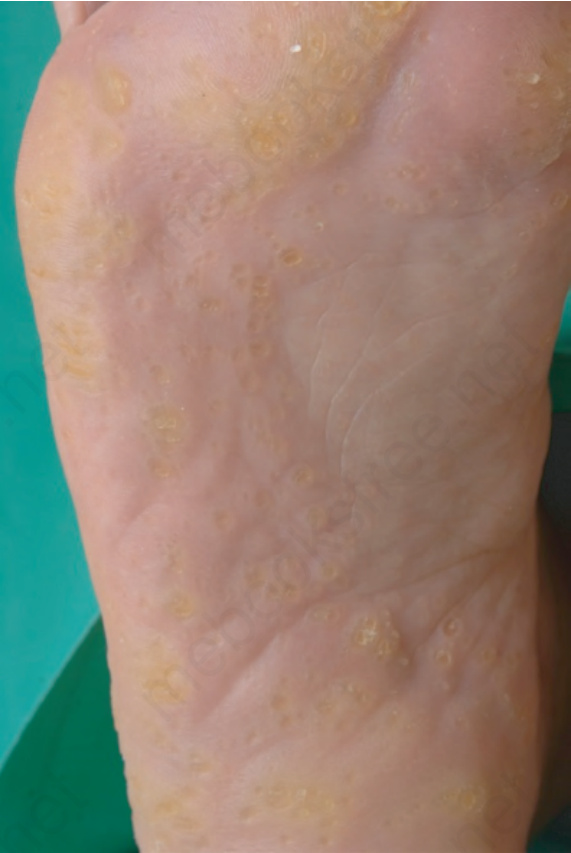
Fig. 3.124 Punctate palmoplantar keratoderma: discrete yellow keratotic papules over pressure areas coalesce into larger plaques.
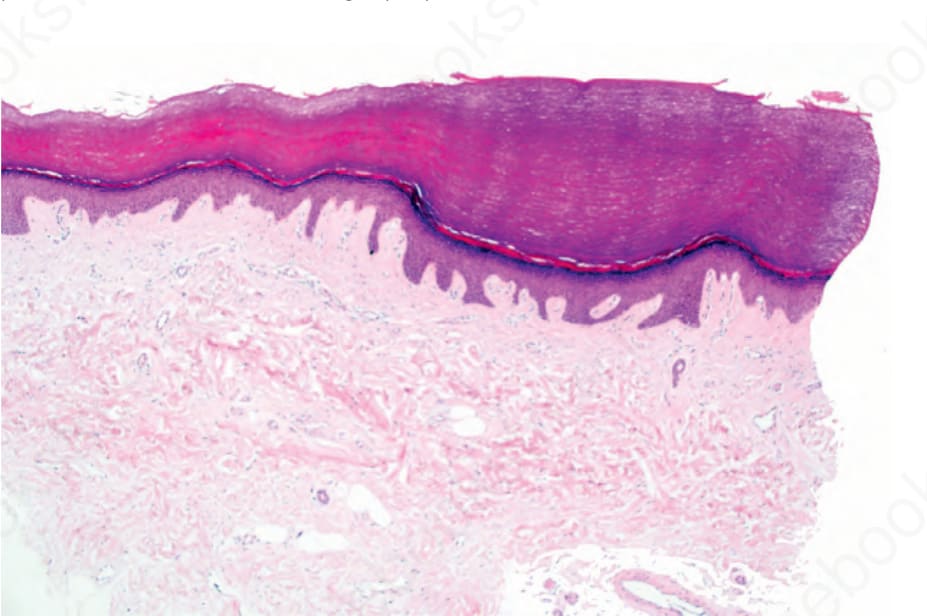
Fig. 3.125 Punctate palmoplantar keratoderma: there is massive orthohyperkeratosis overlying an epidermal depression.
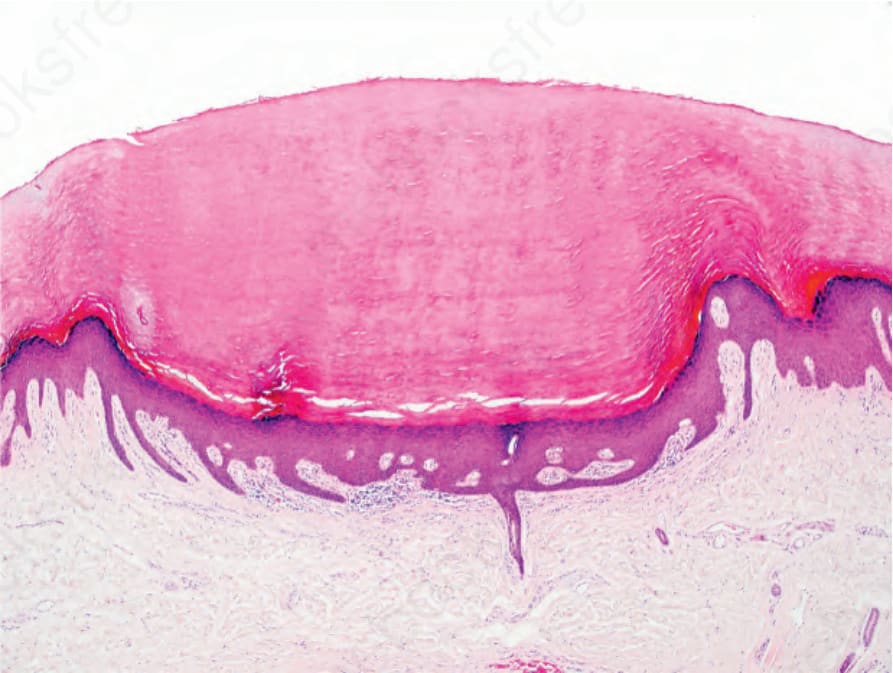
Fig. 3.126 Punctate palmoplantar keratoderma: elongated and curved rete ridges may be seen and are often misdiagnosed as verruca vulgaris.