臨床特徵 (Clinical Features)
- McGrath 症候群 (McGrath syndrome,又稱外胚層發育不良-皮膚脆弱症候群 (ectodermal dysplasia-skin fragility syndrome)) 以體染色體隱性 (autosomal recessive) 模式遺傳,最適合歸類於表皮鬆解水疱症 (epidermolysis bullosa) 之範疇。
- 新生兒呈現紅色脫皮的皮膚 (peeling reddish skin) 與足底水疱 (blisters on the soles)。其後病人發展出瀰漫性、有時呈疣狀 (verruciform) 的掌蹠過度角化 (palmoplantar hyperkeratosis),伴隨疼痛性龜裂 (painful cracking)、皮膚糜爛 (skin erosions),包括口周裂隙 (perioral fissures),以及其他外胚層發育 (ectodermal development) 異常,例如生長遲緩 (growth delay)、毛髮稀少 (hypotrichosis) 或禿髮 (alopecia)、汗腺分泌減少 (hypohidrosis) 與甲營養不良 (nail dystrophy)。
- 與某些其他遺傳性胞橋小體 (desmosomes) 疾病不同,本症沒有心臟病變 (no cardiac pathology)。
致病機轉與組織學特徵 (Pathogenesis & Histologic Features)
- 本病已被證實與 plakophilin-1 基因 (PKP1) 突變有關,導致 plakophilin 1 完全消失;plakophilin 1 負責將胞橋小體蛋白 (desmosomal proteins) 招募至細胞質膜 (plasma membrane) 並負責與角蛋白 (keratin) 的交互作用。
- 皮膚的光學顯微鏡檢查 (light microscopy) 顯示表皮增厚 (thickening of the epidermis) 與角質細胞間隙廣泛增寬 (extensive widening of keratinocyte intercellular spaces),導致裂隙形成 (clefting) 與水疱 (blistering)。
- 亦可發現程度不一的角質細胞角化不良 (variable dyskeratosis of keratinocytes)。
- plakophilin-1 的皮膚免疫染色 (immunostaining) 完全缺如。
- 取自毛髮稀少 (hypotrichotic) 頭皮的切片顯示退化-休止期毛囊 (catagen-telogen hair follicles) 數量增加。
- 電子顯微鏡 (electron microscopy) 顯示角質細胞與角質細胞間黏附 (keratinocyte-keratinocyte adhesion) 喪失。胞橋小體 (desmosomes),特別是位於下方棘上層 (lower suprabasal layers) 者,體積小且數量減少。胞橋小體的內、外斑塊 (inner and outer desmosomal plaques) 發育不良。
- 角化不良的角質細胞 (dyskeratotic keratinocytes) 顯示角蛋白絲 (keratin filaments) 自胞橋小體脫離,並伴有核周凝聚 (perinuclear condensation)。
註:以下段落於原始 OCR 檔中與 McGrath syndrome 內容混置,依「圖文完整保留、不重新歸屬」之鐵則,全文保留並譯出。
-
掌蹠角化 (keratoderma) 為條紋狀 (striated) 而非如 Naxos disease 中通常所見的瀰漫狀 (diffuse)。最早出現的心臟異常僅見於心電圖 (electrocardiographic),且發生於無症狀的病人。在這些病人中,左心室擴大 (dilatation of the left ventricle) 連同肌肉收縮力 (muscle contractility) 的改變,可能導致鬱血性心衰竭 (congestive heart failure),並在較年輕時即有高死亡率 (high mortality rate)。
-
致病機轉與組織學特徵 (Pathogenesis and histologic features):Carvajal-Huerta syndrome 由突變的 desmoplakin 1 (DSP) 所引起;desmoplakin 是胞橋小體的主要組成成分,負責表皮與心臟組織的剛性與強度 (rigidity and strength)。desmoplakin 的其他基因缺陷已被發現可產生廣泛的表現型,其中有些缺乏皮膚症狀,有些則缺乏心臟異常。desmoplakin 的殘餘量對於維持表皮完整性至關重要,如同合子複合突變 (compound heterozygote) 病人攜帶一個無效對偶基因 (null-allele) 與一個錯義突變 (missense mutation) 者所示,此類病人發展出明顯的皮膚脆弱 (skin fragility) 與禿髮 (alopecia) 而無心臟異常。desmoplakin 尾域 (tail domain) 完全喪失者,則表現為「致死型」棘層鬆解性表皮鬆解水疱症 (‘lethal’ acantholytic epidermolysis bullosa)。
-
組織學顯示正角化性過度角化 (orthohyperkeratosis)、細胞間隙增寬 (widening of intercellular spaces),以及棘上層角質細胞 (suprabasal keratinocytes) 的部分裂解 (partial dehiscence)(圖 3-116)。此特徵性型態的組織學鑑別診斷見表 3-8。
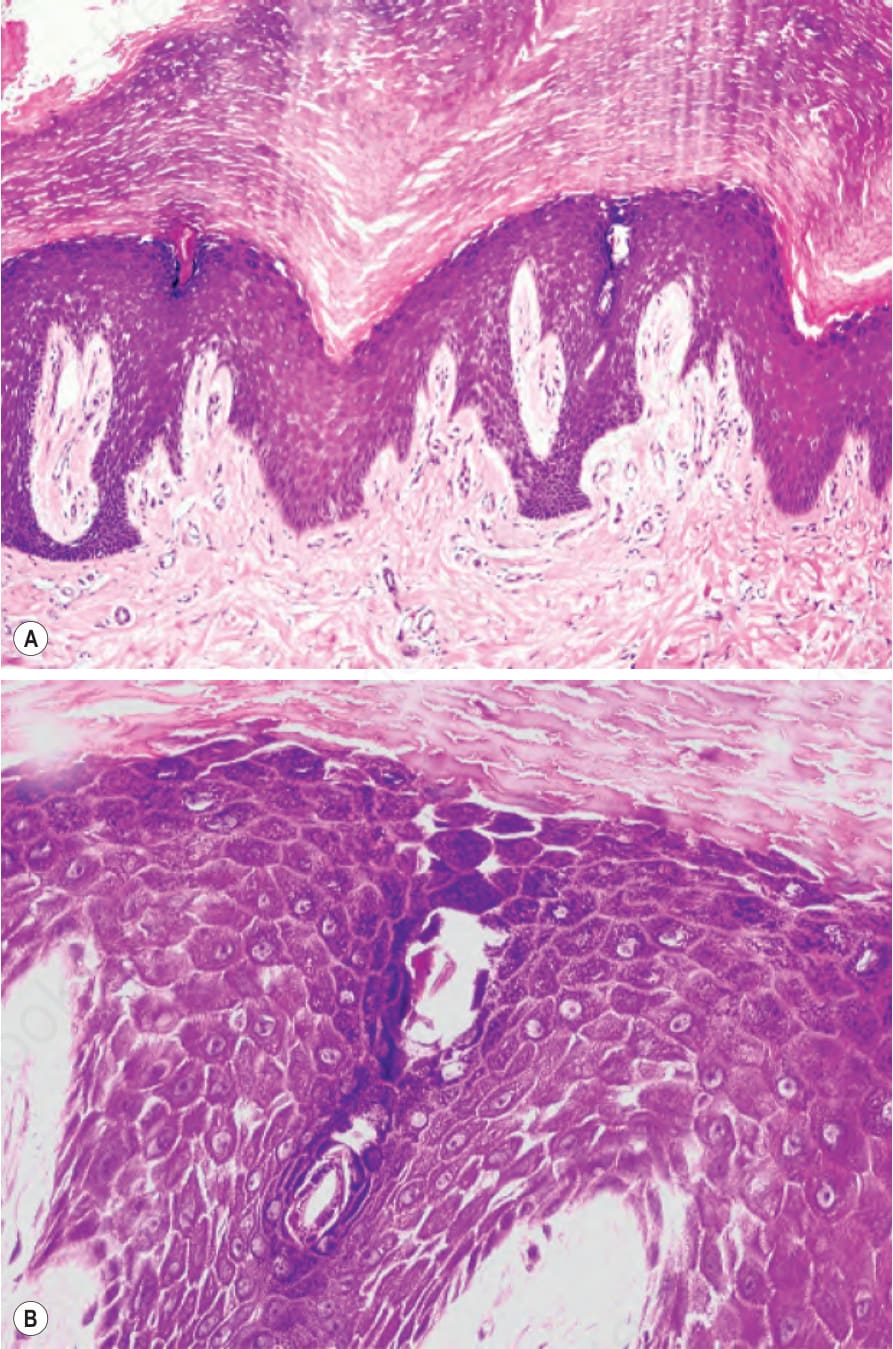
圖 3-114:條紋狀掌蹠角化症 (striate palmoplantar keratoderma):(A) 組織學顯示大量正角化性過度角化 (massive orthohyperkeratosis)、顆粒層增厚 (hypergranulosis) 與棘層肥厚 (acanthosis);(B) 角質細胞間黏聚力喪失導致特徵性的細胞間隙增寬 (widening of the intercellular spaces)。
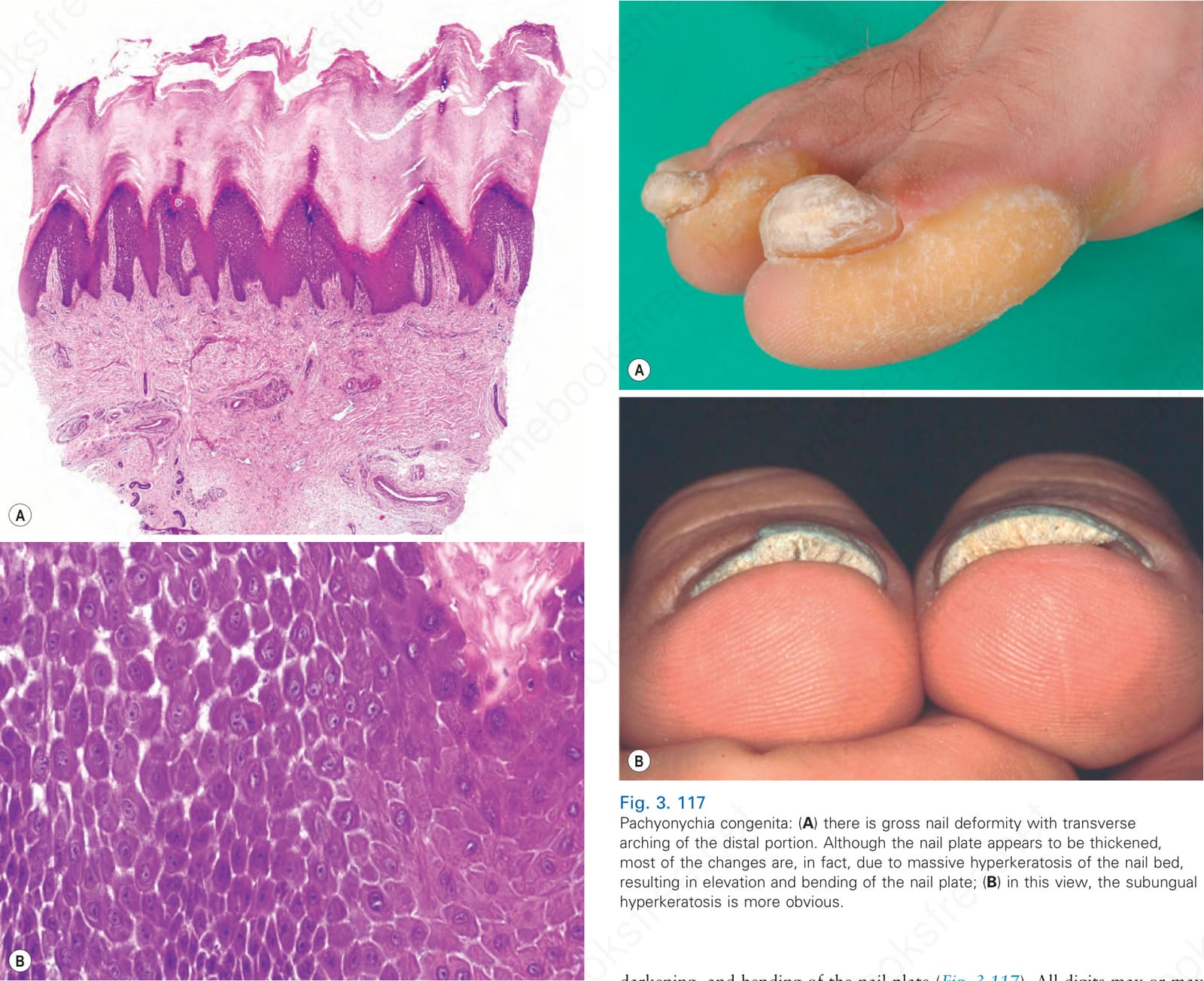
圖 3-116:Carvajal-Huerta syndrome:(A) 大量正角化性過度角化 (massive orthohyperkeratosis)、棘層肥厚 (acanthosis) 與乳頭瘤狀增生 (papillomatosis);(B) 棘上層角質細胞的部分裂解 (partial dehiscence of suprabasal keratinocytes) 與特徵性的細胞間隙增寬 (widening of the intercellular spaces)。
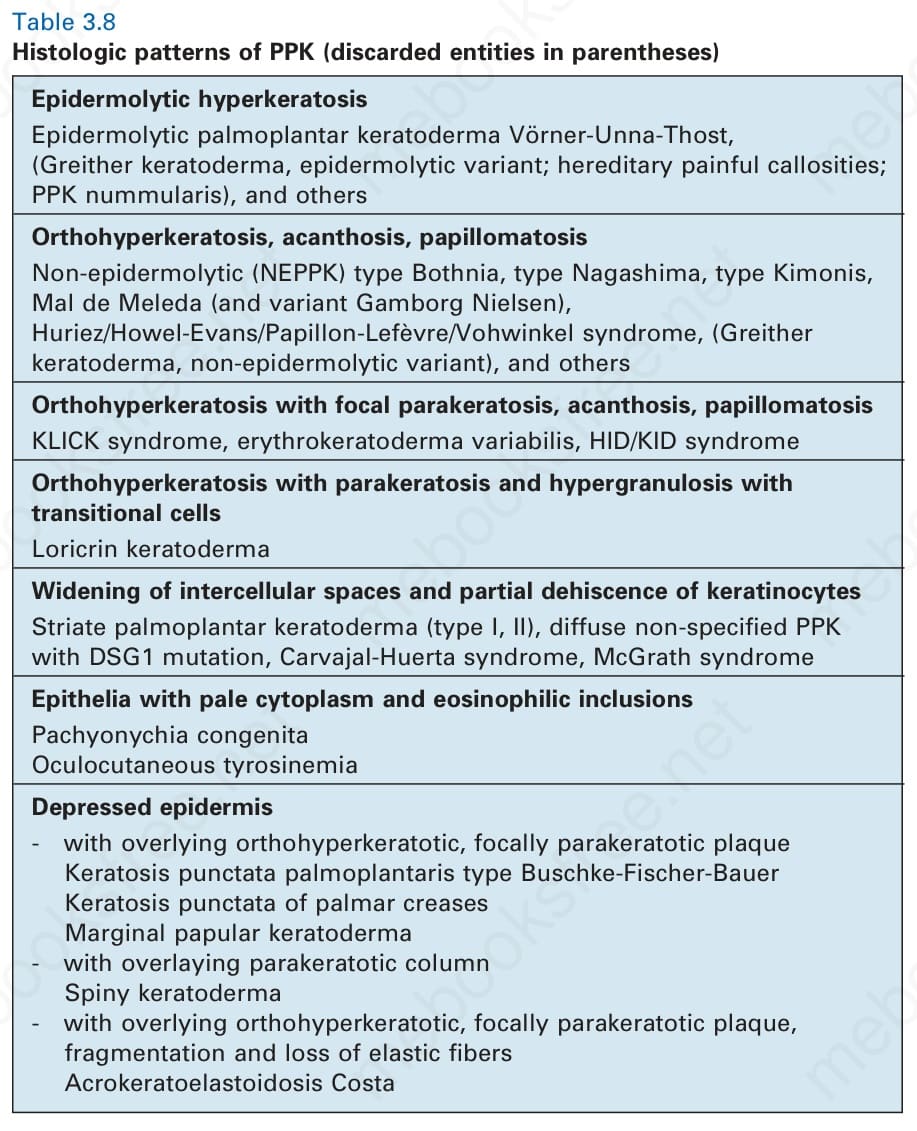
表 3-8:PPK 的組織學型態 (histologic patterns of PPK,已廢棄的疾病名稱以括號標示)。