McGrath syndrome
McGrath syndrome
Clinical features McGrath syndrome (ectodermal dysplasia-skin fragility syndrome) is inherited in an autosomal recessive mode and is best classified in the category of epidermolysis bullosa. Newborns present with peeling reddish skin and blisters on the soles. Thereafter, patients develop diffuse, sometimes verruciform palmoplantar hyperkeratosis with painful cracking, skin erosions, including perioral fissures, and other abnormalities of ectodermal development, such as growth delay, hypotrichosis or alopecia, hypohidrosis, and nail dystrophy.3 In contrast to some other inherited disorders of desmosomes, there is no cardiac pathology.
B
keratoderma is striated rather than diffuse as usually occurring in Naxos disease. The first cardiac abnormalities are exclusively electrocardiographic and occur in asymptomatic patients. In these patients, dilatation of the left ventricle, together with alterations in muscle contractility, may lead to congestive heart failure with a high mortality rate at younger age.3
Pathogenesis and histologic features The disease has been shown to be associated with mutations in the plakophilin-1 gene (PKP1) leading to complete ablation of plakophilin 1, which is responsible for recruitment of desmosomal proteins to the plasma membrane and for keratin interaction.1,2,4
Light microscopy of the skin shows thickening of the epidermis and extensive widening of keratinocyte intercellular spaces leading to clefting and blistering.1,2 Variable dyskeratosis of keratinocytes may also be found.5 There is complete absence of cutaneous immunostaining for plakophilin-1.4 Biopsies from the hypotrichotic scalp demonstrate increased number of catagen-telogen hair follicles.5 Electron microscopy reveals loss of keratinocyte–keratinocyte adhesion. Desmosomes, particularly in the lower suprabasal layers, are small and reduced in number. The inner and outer desmosomal plaques are poorly developed.6 Dyskeratotic keratinocytes reveal detachment of the keratin filaments from the desmosomes with perinuclear condensation.5
Pathogenesis and histologic features Carvajal-Huerta syndrome is caused by mutant desmoplakin 1 (DSP), a major constituent of desmosomes that is responsible for the rigidity and strength of the epidermis and cardiac tissue.4,5 Other genetic defects in desmoplakin have been found to generate a wide range of phenotypes, some of them lacking cutaneous symptoms, others lacking cardiac anomalies.6 The residual amount of desmoplakin is critical in maintaining epidermal integrity as illustrated by compound heterozygote patients carrying one null-allele and one missense mutation, who developed pronounced skin fragility and alopecia without cardiac anomalies.7 Complete loss of the tail domain of desmoplakin presents as ‘lethal’ acantholytic epidermolysis bullosa.8
Histology shows orthohyperkeratosis and widening of intercellular spaces and partial dehiscence of suprabasal keratinocytes (Fig. 3.116). For the histologic differential diagnosis of this characteristic pattern see Table 3.8. At
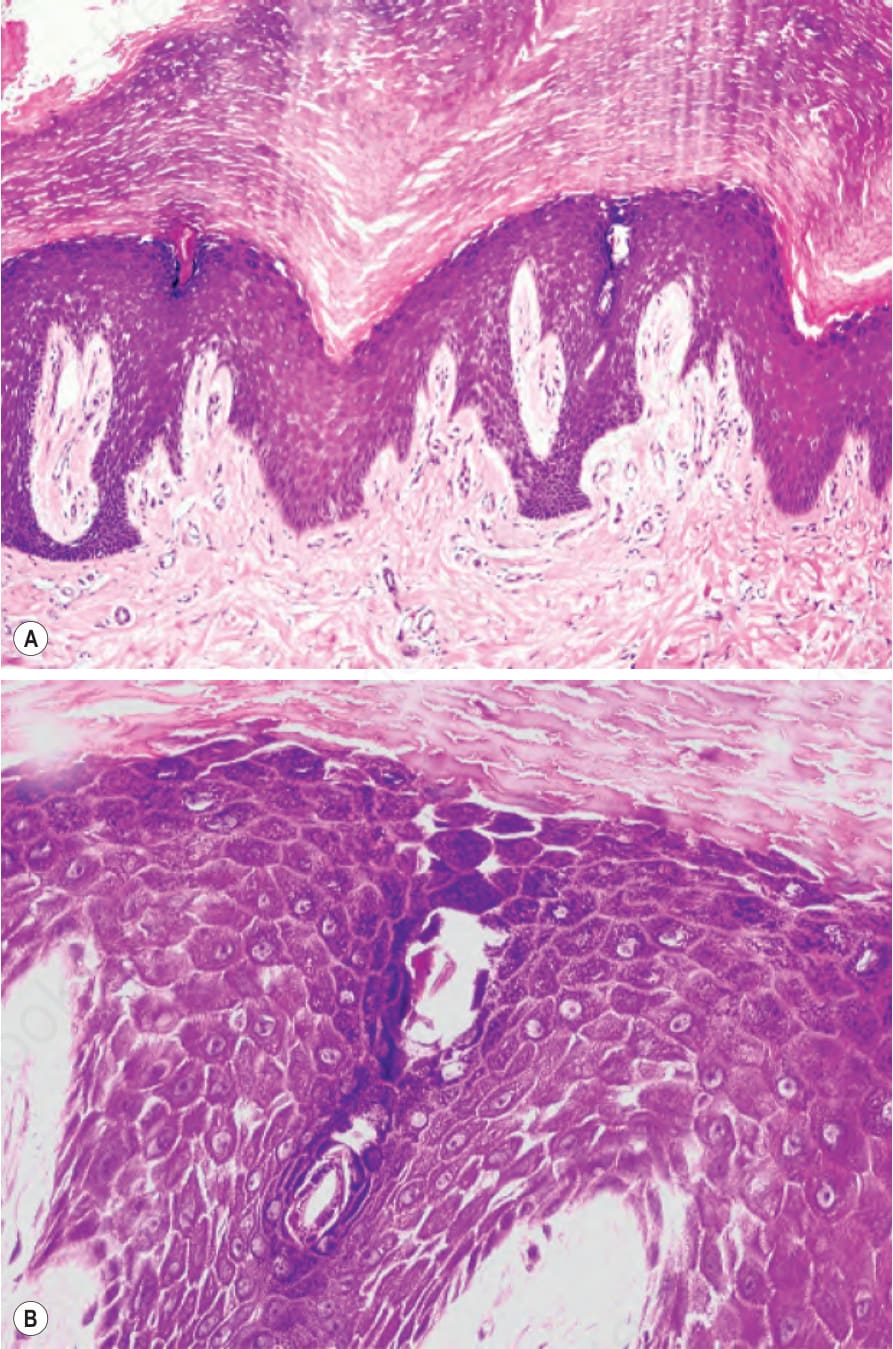
Fig. 3.114 Striate palmoplantar keratoderma: (A) histology reveals massive orthohyperkeratosis, hypergranulosis, and acanthosis; (B) loss of cohesion of keratinocytes leads to characteristic widening of the intercellular spaces.
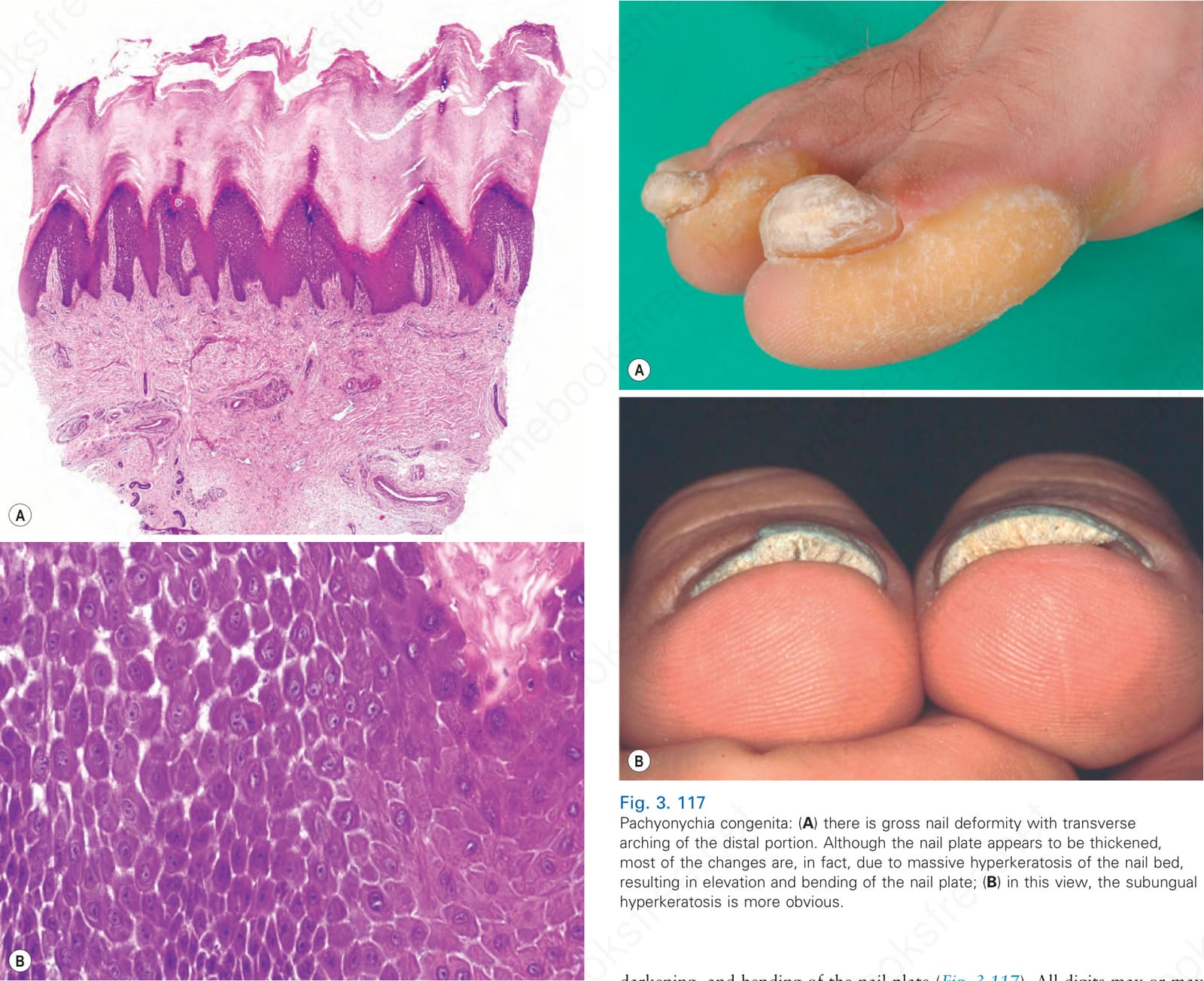
Fig. 3.116 Carvajal-Huerta syndrome: (A) massive orthohyperkeratosis, acanthosis, and papillomatosis; (B) partial dehiscence of suprabasal keratinocytes and characteristic widening of the intercellular spaces.
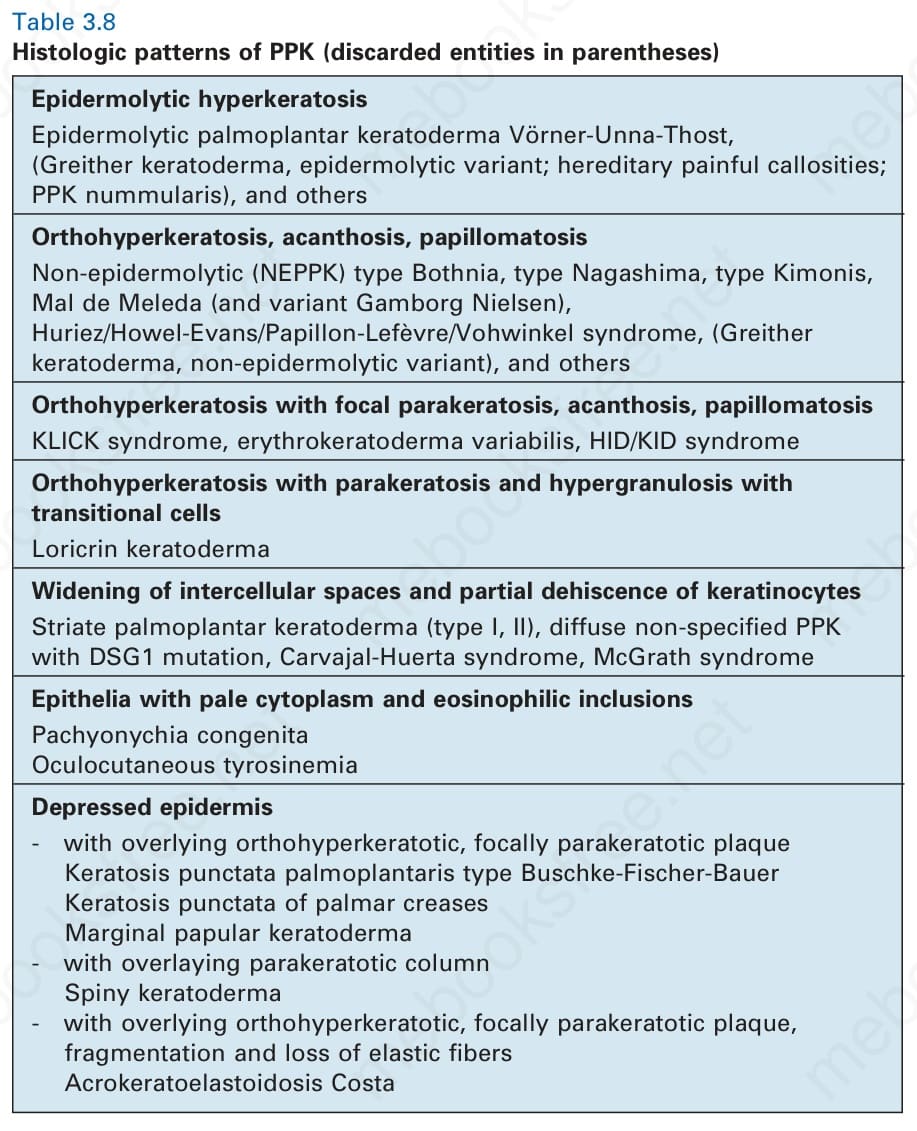
Table 3.8 Histologic patterns of PPK (discarded entities in parentheses)