次世代定序 (Next-generation sequencing, NGS)
大規模平行定序 (massively parallel) 或次世代定序 (next-generation sequencing, NGS) 近年來已徹底改變了研究與診斷兩大領域。它使許多癌症的基因組樣貌 (genomic landscapes) 得以被詳盡描述,並協助分離出許多孟德爾遺傳性疾病 (Mendelian heritable diseases) 的致病基因。
NGS 是一組極具彈性的技術與方法,可加以調整以:
- 定序整個基因組、外顯子體 (exome) 或其任一子集(DNA)中的變異,
- 提供 DNA 拷貝數資訊,
- 定序整個轉錄體 (transcriptome,即被轉錄的 RNA) 或其任一子集,
- 辨識易位 (translocations),
- 呈現基因表現量,
- 使用新鮮或 FFPE 材料,
- 但確實需要複雜的生物資訊學 (bioinformatic) 判讀,並具有龐大的資料儲存需求。
NGS 使用雜交捕捉 (hybrid capture) 或以擴增子為基礎 (amplicon based) 的技術來定序 DNA 或 RNA(先前反轉錄為 cDNA)。其範圍可從整個基因組(全部約 30 億個鹼基對 (~3 billion base pairs)),到所有編碼基因(外顯子體;約佔基因組的 1% 或約 3000 萬個鹼基對 (~30 million base pairs)——即約 20,000 個基因,由 180,000 個外顯子組成),到所有由基因轉錄而來的 RNA(轉錄體),以及這些的任一子集。因此它在容量上大幅超越先前的方法,並很可能在可預見的未來推動分子檢測的發展。此技術使用片段化的 DNA 來建構定序文庫 (sequencing library),隨後同時定序數百萬條個別的 DNA 股。視所採用的方法而定,可使用不同的化學原理,但全部皆使用矽微晶片 (silicon microchip) 以同步記錄序列。所有測定出的序列隨後以複雜的電腦演算法,比對一個公認的參考基因組 (reference genome) 進行排比,以辨識差異。由於個別被定序的 DNA 片段是分開排比的,故突變的量化(等位基因頻率 (allelic frequencies))可被非常精確地測定。當在癌症中進行廣泛定序(超出僅僅少數幾個基因)時,常有必要定序自淋巴球或其他正常組織萃取的正常(非腫瘤)DNA,以資比較並辨識出腫瘤過程所特有的變化。
首個被定序的人類基因組需動用全球 20 個卓越中心的數百名工作人員,耗資 27 億美元 (1000),基本上由單一人員即可完成——不過徹底的判讀則複雜得多。在臨床實務中,通常檢測較小的基因組,範圍從約 10 個到數百個。雖然真正具臨床可採取行動性 (clinically actionable) 的基因僅有少數幾個,但檢視較大的基因組合有助於癌症的分子分類 (molecular classification),並為臨床試驗的選擇提供指引。已有眾多資料庫可協助判讀可能非常複雜的定序結果,並決定哪些結果可能具有臨床意義。使用公認且明確的命名法 (unambiguous nomenclature) 對於溝通癌症與遺傳性疾病的定序結果亦相當重要。
NGS 的應用無疑將以可預期、或許也以未曾預期的方式持續成長。
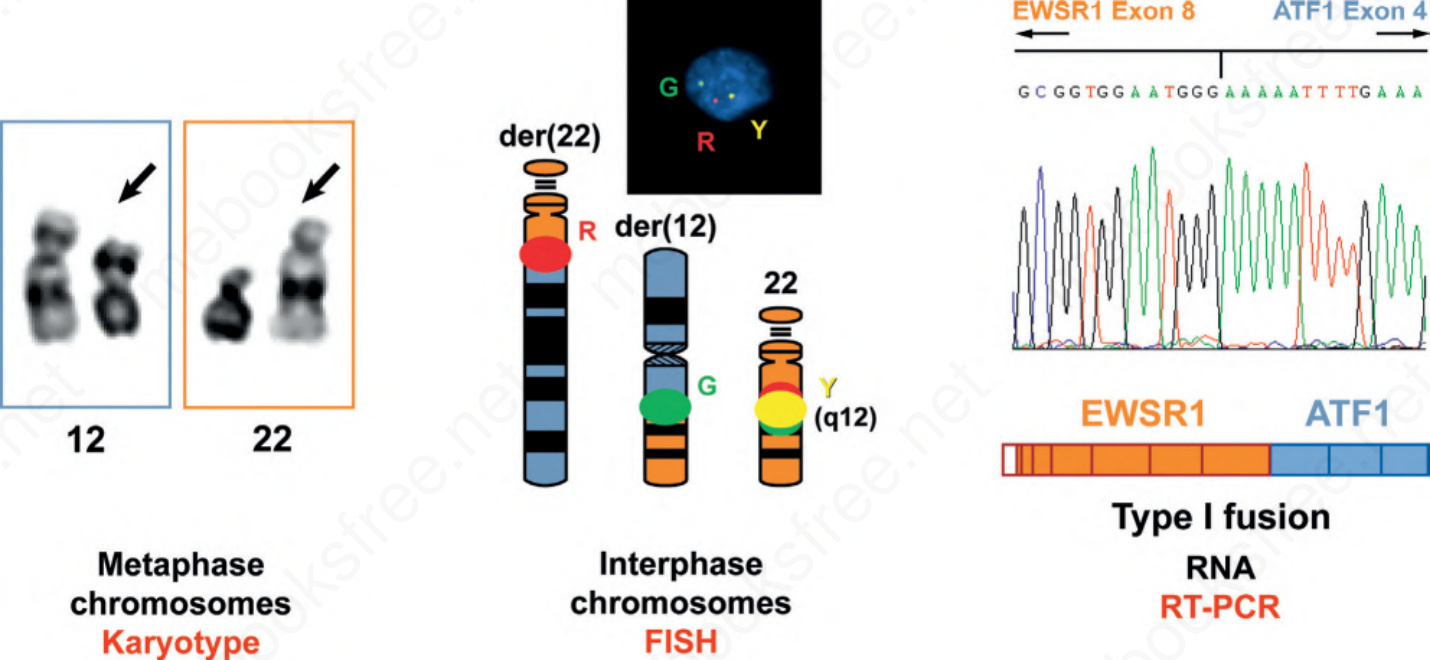
圖 2-30:偵測反覆性易位的多種模式。傳統核型分析使用中期染色體展開,藉由分帶(染色)技術偵測易位及其他結構性遺傳異常。FISH 使用較不凝縮的間期染色體以偵測重排或擴增。RT-PCR 可偵測融合 RNA 轉錄本中所涉的精確外顯子。每一種皆為呈現染色體易位的有效方法,但各自適用於不同的樣本類型並提供不同的資訊。

圖 2-31:整合式基因組檢視器 (integrated genome viewer, IGV) 對 melanoma 中一個 BRAF V600E 突變的呈現。請注意,此顯示展示了由 NGS 定序在雙向(正股與反股,藍色與紅色)所定序的個別 DNA 股。因此可測定精確的變異等位基因頻率 (variant allele frequency)。