Next-generation sequencing (NGS)
Next-generation sequencing (NGS)
Massively parallel or next-generation sequencing (NGS) has dramatically altered both the research and diagnostic arenas in recent years. It has allowed detailed descriptions of the genomic landscapes of many cancers and helped to isolate the causative gene in many Mendelian heritable diseases.29–31
50 Specialized techniques in dermatopathology
NGS is an exceedingly flexible set of techniques and approaches and can be adapted to:
• sequence alterations in the entire genome, exome, or any subset thereof (DNA),
• provide DNA copy number information,
• sequence the entire transcriptome (transcribed RNA) or any subset thereof,
• identify translocations,
• demonstrate gene expression levels,
• use fresh or FFPE material,
• but does require complex bioinformatic interpretation and has extensive data storage requirements. NGS uses hybrid capture or amplicon based techniques to sequence DNA or RNA (previously transcribed to cDNA). This can range from the entire genome (all ~3 billion base pairs), to all coding genes (exome; ~1% of the genome or ~30 million base pairs—that is ~20,000 genes composed of 180,000 exons), to all of the RNA transcribed from genes (transcriptome) and any subset of these. Thus it greatly outpaces prior approaches in capacity and will likely fuel molecular testing for the foreseeable future.32 The technique uses fragmented DNA to construct a sequencing library, and then millions of individual DNA strands are sequenced simultaneously. Different chemistries can be employed depending on the approach, but all use a silicon microchip for simultaneous recording of sequences. All the determined sequences are then aligned using complex computer algorithms against an accepted reference genome to identify differences. Because individual sequenced DNA segments are aligned separately, quantification of mutations (allelic frequencies) can be very precisely determined. When extensive sequencing (beyond just a few genes) is done in cancer, it is often necessary to sequence normal (non-neoplastic) DNA extracted from lymphocytes or other normal tissue to compare and identify the changes specific to the neoplastic process.
The first human genome sequenced required many hundreds of workers at 20 centers of excellence around the world, cost 1000, essentially by a single person—though a thorough interpretation will be much more involved. In clinical practice, smaller genes sets are usually interrogated ranging from around 10 to several hundred.34 While only a handful of genes are truly clinically actionable, examination of larger gene sets can aid the molecular classification of cancer and provide guidance for the selection of clinical trials.35 Numerous databases are available to help interpret sequencing results, which can be very complex, and decide which results are likely to be clinically meaningful.36 Using accepted unambiguous nomenclature is also important to communicate sequencing results for both cancer and heritable diseases.37,38
of NGS will undoubtedly grow in both expected and perhaps unantici pated ways.
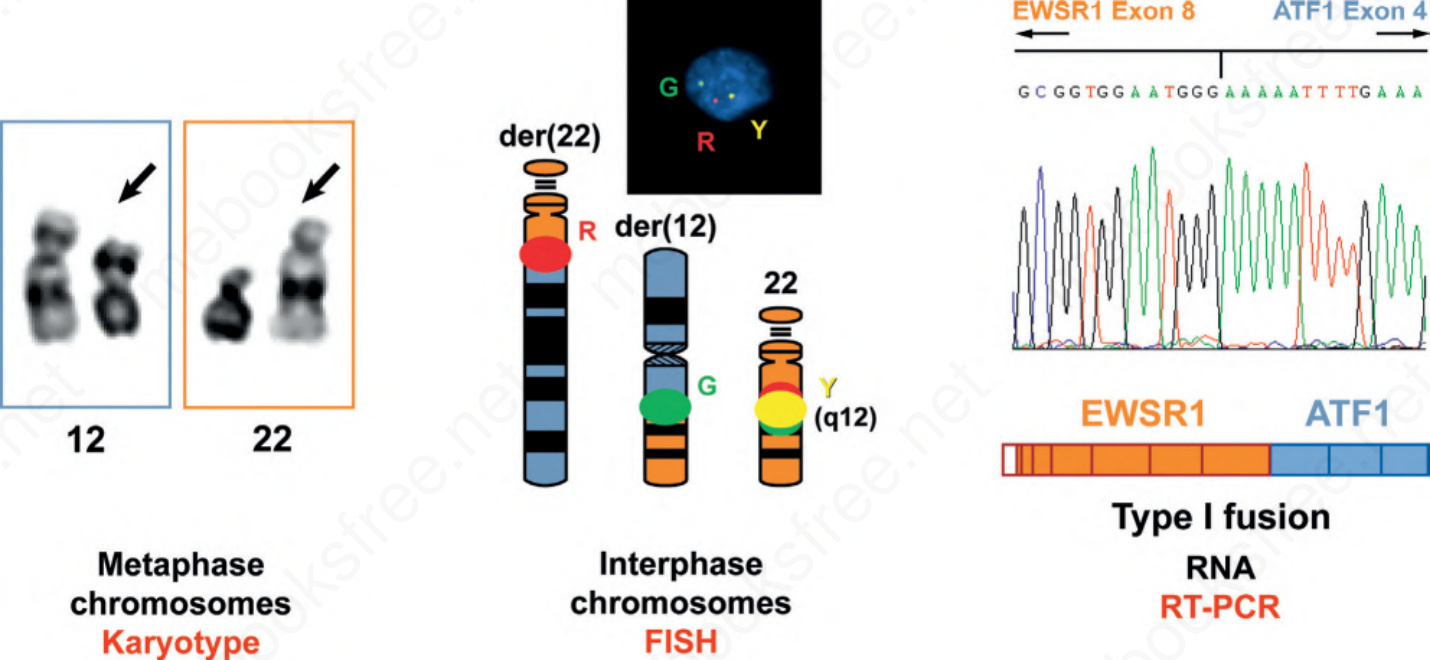
Fig. 2.30 Multiple modalities for detection of recurrent translocations. Traditional karyotypes use metaphase chromosomes spreads to detect translocations and other structural genetic aberrations using banding (staining) techniques. FISH uses less condensed interphase chromosomes to detect rearrangements or amplifications. RT-PCR can detect the precise exons involved in a fusion RNA transcript. Each is a valid method for demonstrating chromosomal translocations, but each has applicability to different sample types and provides different information.

Fig. 2.31 Integrated genome viewer (IGV) depiction of a BRAF V600E mutation in melanoma. Note that the display shows individual DNA strands sequenced in both directions (forward and reverse strands, blue and red) by NGS sequencing. Thus the precise variant allele frequency can be determined.