Neurofibromatosis
Neurofibromatosis
Clinical features Traditionally neurofibromatosis is classified into:
• the classic peripheral cutaneous variant or type I neurofibromatosis (NF1, von Recklinghausen disease; NF1 gene at 17q11.2),
Neurofibromas presenting at birth are often plexiform and are located particularly around the eyes and neck; some are generalized.26 By late childhood or adolescence, large numbers of cutaneous tumors have developed, which may be nodular, sessile or pedunculated (Figs 35.345–35.349). Neurofibromas in NFI have increased vascularity and this may result in prominent bleeding, particularly during surgical excision of large plexiform variants. The increased vascularity may result from elevated tumor cell expression of basic fibroblast and endothelial growth factors.27
The diffuse neurofibroma which is generally found on the head, neck and back in a proportion of cases is also associated with neurofibromatosis;
1795 Benign neural tumors
1796 Connective tissue tumors
Neurofibromatosis may also be associated with a diverse range of other manifestations including short stature, pheochromocytoma, gastrointestinal neoplasms (including adenocarcinoma, carcinoid, somatostatinoma and gastrointestinal stromal tumor), mental retardation and a variety of central nervous system tumors (mainly low-grade gliomas but also high-grade tumors including medulloblastoma).11,12,29–33 An association with juvenile xanthogranuloma and leukemia in children is also known.34 A few cases of achondroplasia and NF1 have been documented.35 A number of other associations have been recorded but they are likely to be coincidental. These include cutaneous T-cell lymphoma, epidermodysplasia verruciformis, urticaria pigmentosa, piebaldism, eccrine angiomatous hamartoma, segmental unilateral lentiginosis and multiple glomus tumors.36–43
however, in contrast to the plexiform type, it does not appear to be associated with an increased risk of malignant transformation except in rare cases (Figs 35.350 and 35.351).
Interestingly, biopsies from normal skin in patients with NF1 show an increase in the number of S100 protein-positive cells.28
Occasionally, large plexiform neurofibromas may be associated with excessive redundant skin folds, giving rise to the so-called elephantiasiform neurofibroma (Fig. 35.352).
Lisch nodules are pigmented hamartomas of the iris and are pathognomonic of NF1 (Fig. 35.353).12 However, they are never found in the acoustic or segmental variants.
Patients with neurofibromatosis may develop tumors at any site in the body, including internal nerve trunks and viscera (Figs 35.354 and 35.355). As this is a progressive disorder, increasing age is associated with the acquisition of further nodules; ultimately the patient may exhibit sometimes grotesque features with accompanying psychological and social problems.
Multiple glomus tumors of the digits, however, have been identified as an important association of NF1.44,45 An association with Noonan syndrome is also common and both conditions are pathogenetically related.46,47
Factors found in a recent study to be associated independently with mortality in NF1 include the presence of subcutaneous neurofibromas, the absence of cutaneous neurofibromas and facial asymmetry.48 Independent cutaneous predictor factors associated with internal neurofibromas include the presence of at least two subcutaneous neurofibromas, age = or less than 30, absence of cutaneous neurofibromas and fewer than six café-au-lait
1797 Benign neural tumors
spots.49,50 Based on the latter, a scoring system has been proposed to calculate the risk of internal neurofibromas.
Blue–red macules and pseudoatrophic macules in patients with NF1 have been shown to indicate the presence of neurofibromas.51
Plexiform neurofibromas in NF1 may increase in size during pregnancy but tumors do not appear to express progesterone receptors.52,53
As mentioned earlier, patients with NF1 have an increased risk of developing malignant peripheral nerve sheath tumors, with a lifetime incidence of between 8% and 13%.54–59 Patients with NF1 tend to present earlier in life than those with sporadic malignancy.59 They also tend to present with recurrences and metastatic spread at shorter intervals than patients with sporadic tumors.60 Pain and enlargement are the most frequent signs suggesting malignant transformation. Most lesions occur in the limbs; these are highly aggressive tumors and patients have a mean survival of 18 months. Tumor volume and expression of TP53 have been found to be independent factors predictive of poor behavior.61,62 By the use of murine models, it has
been demonstrated that loss of the tumor suppressor PTEN (phosphatase and tensin homolog) combined with overexpression of the KRAS oncogene is crucial in the development of malignant transformation.63,64
Melanocytic differentiation has exceptionally been documented.65
Acoustic neurofibromatosis (NF2) comprises a syndrome of acoustic neuroma (schwannoma; see Fig. 35.277), which is often bilateral, and intracranial and intraspinal neoplasms, including astrocytomas, meningiomas and ependymomas. An exceptional association with a soft tissue perineurioma has been reported.66 The gene for NF2 has been cloned to chromosome 22.67,68
Segmental neurofibromatosis can occur in both NF1 and NF2 as a result of somatic mosaicism.69–74 Segmental neurofibromatosis may occur in a patient with classic NF1 or, more commonly, in patients with no signs of neurofibromatosis other than café-au-lait spots. Most cases of segmental neurofibromatosis have no positive family history.75,76 Involvement is usually unilateral but may be bilateral.74
1798 Connective tissue tumors
Pathogenesis and histologic features Neurofibromatosis type 1 results from gene mutations in the NF1 gene. The NF1 gene is located on chromosome 17q11.2, which encodes for the protein neurofibromin. The molecular mechanisms involved in tumorigenesis remain largely unknown; a plethora of mutations of the NF1 gene have been investigated. Neurofibromatosis type 2 is driven by loss of function of the NF2 gene (22q12.2) encoding the protein merlin (see also schwannoma).77–91
The histologic appearances of the skin and subcutaneous tumors seen in neurofibromatosis have been described under previous headings. Hypertrophy of pacinian corpuscles has been reported in a patient with NF1.92 Increased cellularity and cytologic atypia is found in about one-fifth of cases and may represent an indicator of increased risk of malignant transformation.93 Although it has been reported that floret-like giant cells are associated with tumors in neurofibromatosis type 1, they can also be identified in sporadic neurofibromas.94,95 Malignant peripheral nerve sheath tumors tend to be more cellular but less pleomorphic than sporadic tumors.60
The café-au-lait macules show increased numbers of functionally active melanocytes with giant melanosomes.
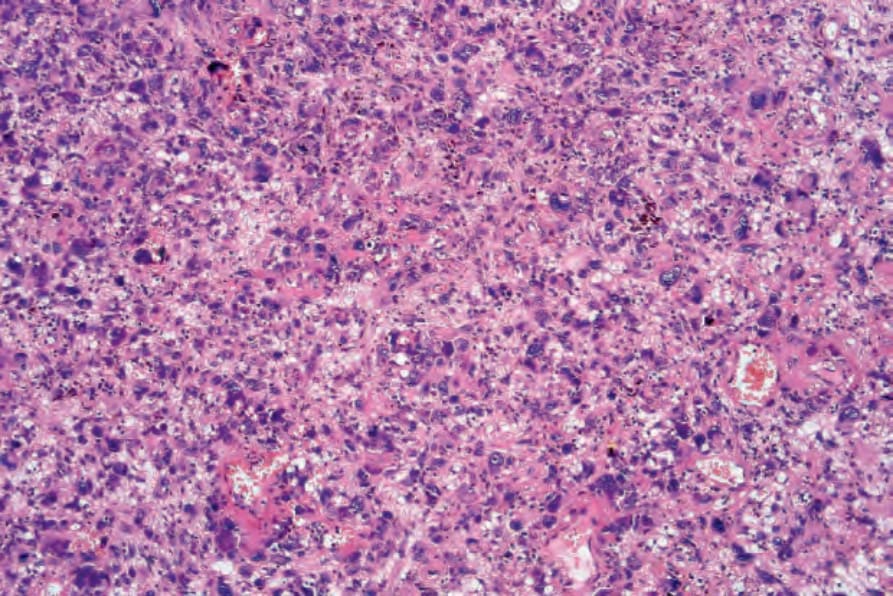
Fig. 35.277 Atypical fibroxanthoma: medium-power view showing a highly cellular and pleomorphic tumor cell infiltrate.
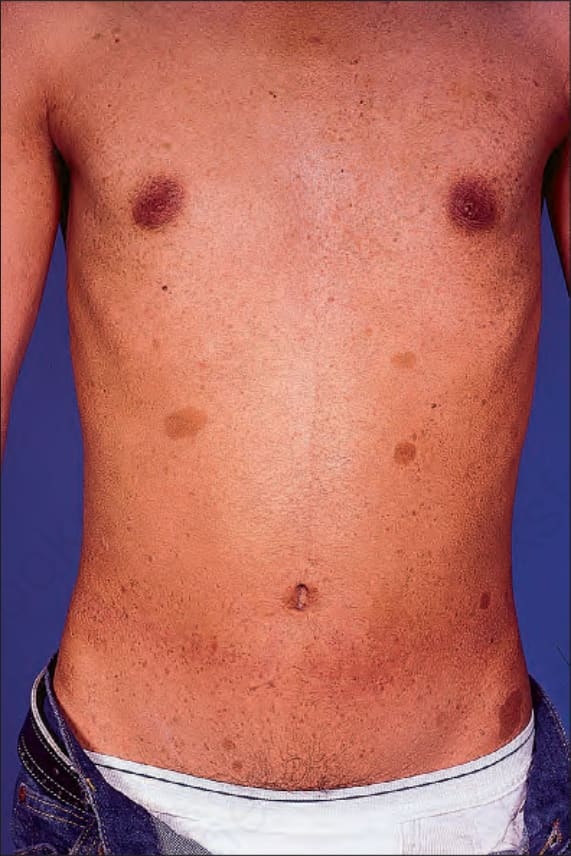
Fig. 35.343 Type I neurofibromatosis: the presence of typical café-au-lait macules is characteristic. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
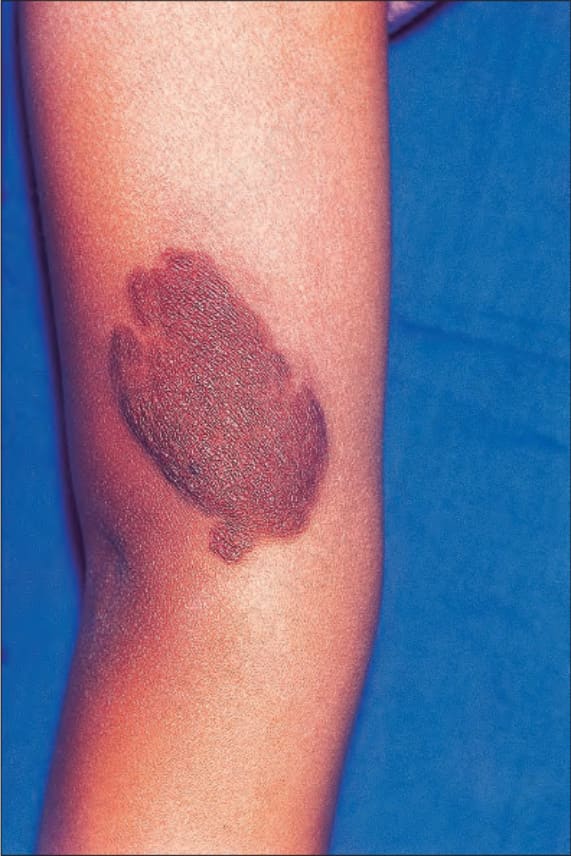
Fig. 35.344 Type I neurofibromatosis: this heavily pigmented raised lesion overlies a cutaneous plexiform neurofibroma. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
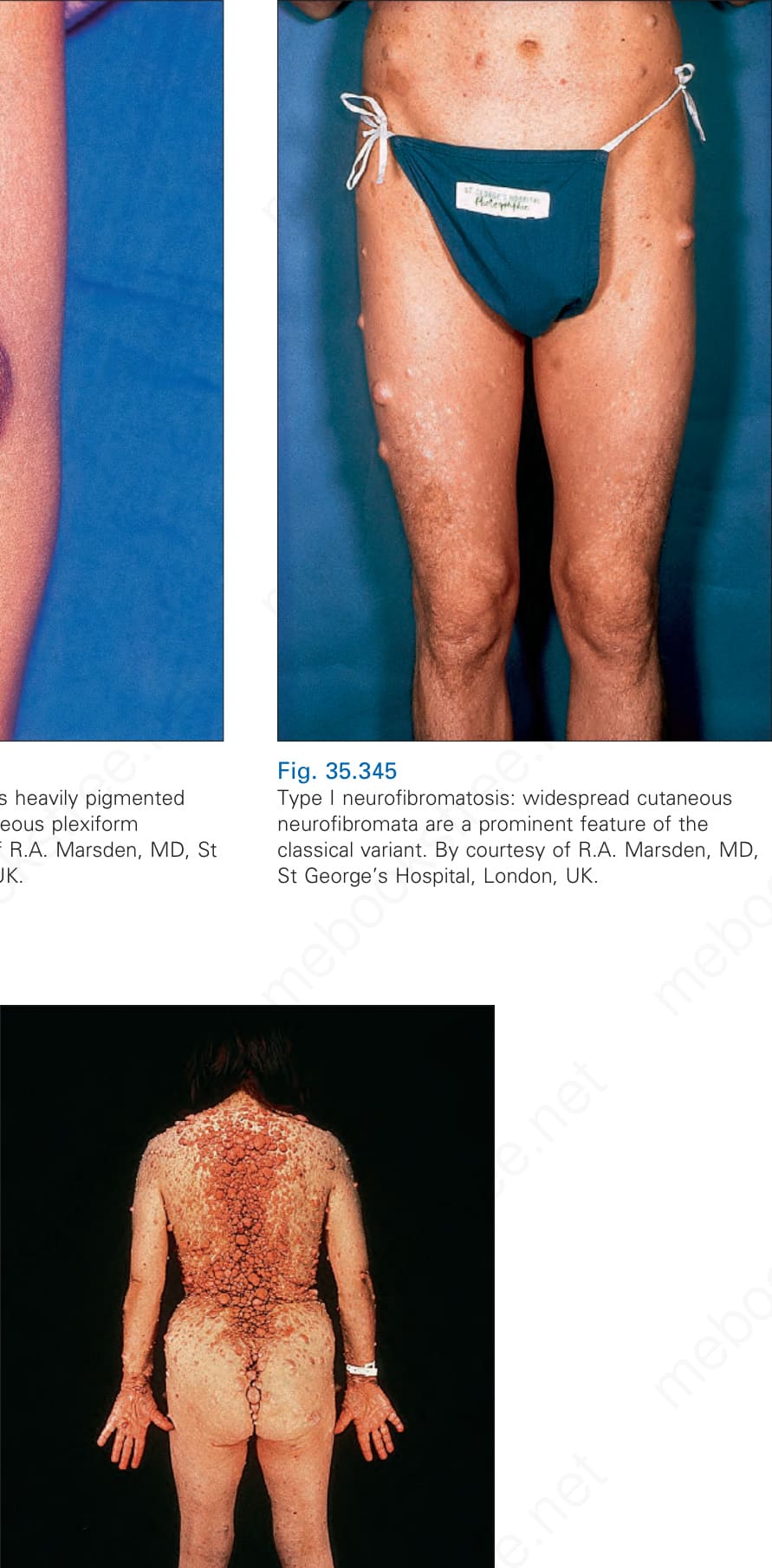
Fig. 35.345 Type I neurofibromatosis: widespread cutaneous neurofibromata are a prominent feature of the classical variant. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
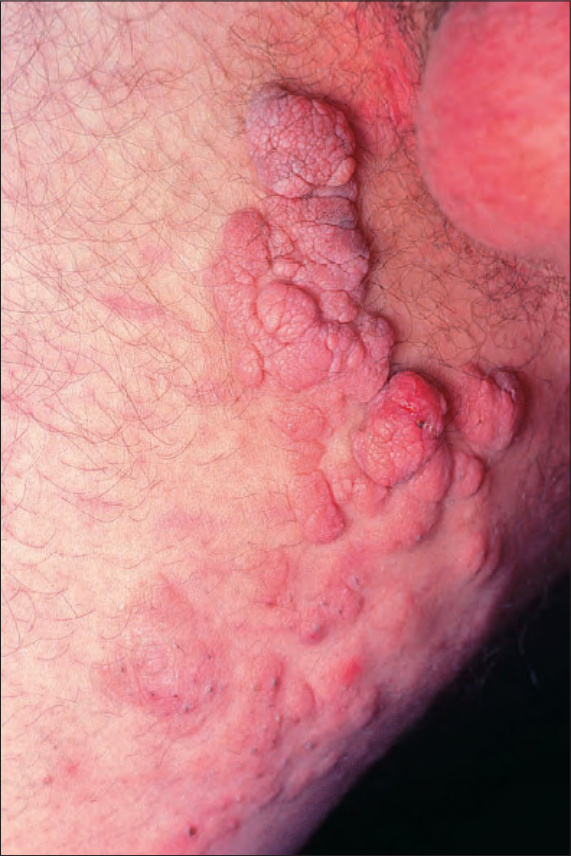
Fig. 35.346 Type 1 neurofibromatosis: lesions are often soft and appear as polypoid or sessile papules and plaques. By courtesy of the Institute of Dermatology, London, UK.
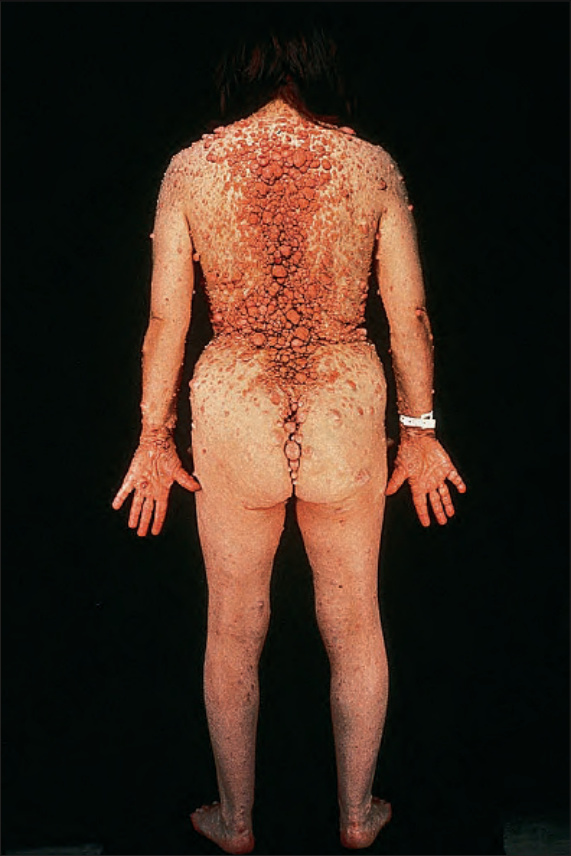
Fig. 35.347 Type I neurofibromatosis: this disease can be extremely disfiguring. By courtesy of the Institute of Dermatology, London, UK.
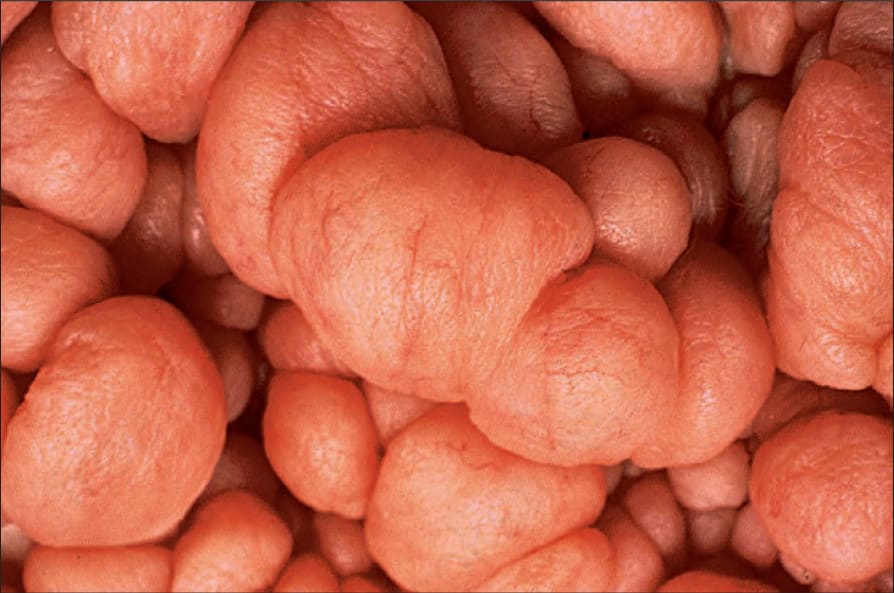
Fig. 35.348 Type 1 neurofibromatosis: close-up view. By courtesy of the Institute of Dermatology, London, UK.
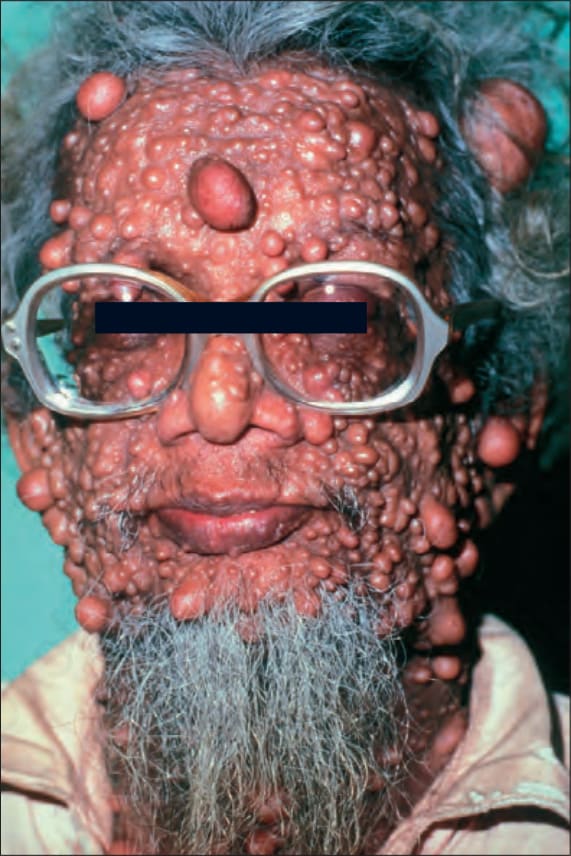
Fig. 35.349 Type 1 neurofibromatosis: there is marked facial disfigurement. By courtesy of the Institute of Dermatology, London, UK.
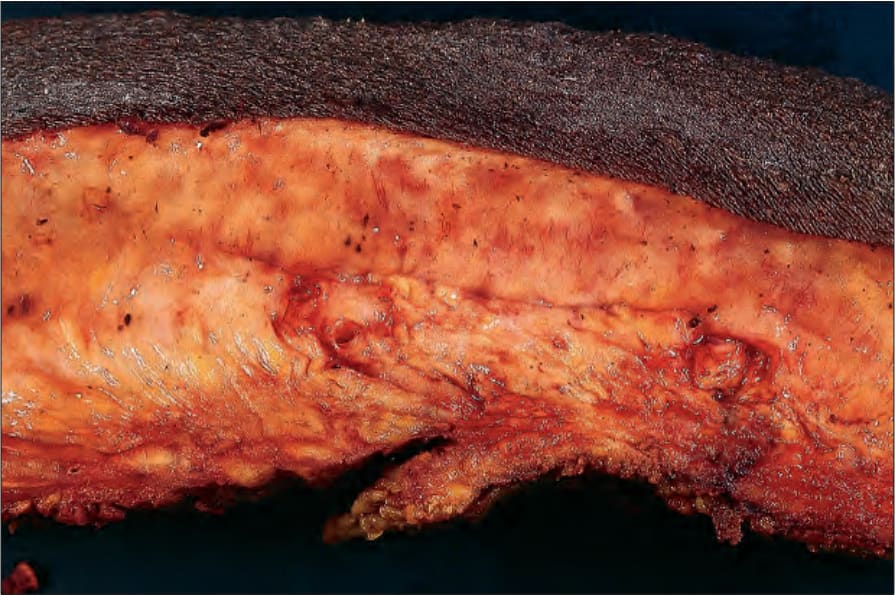
Fig. 35.350 Type I neurofibromatosis: there is extensive replacement of dermis and subcutaneous fat by a homogeneous pale yellow tumor – a diffuse neurofibroma.

Fig. 35.351 Type 1 neurofibromatosis: the skin overlying the tumor has a wrinkled, unevenly elevated appearance.

Fig. 35.352 Type I neurofibromatosis: the elephantiasiform variant. By courtesy of D. Allen, MD, St Thomas’ Hospital, London, UK.
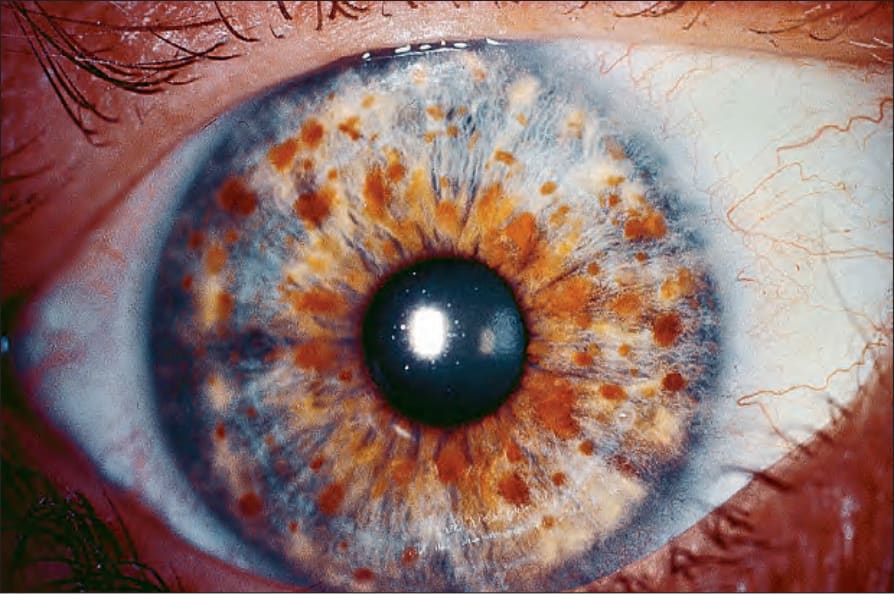
Fig. 35.353 Type I neurofibromatosis: multiple Lisch nodules (iris nevi) are a pathognomonic feature. By courtesy of D. Spalton, MD, St Thomas’ Hospital, London, UK.
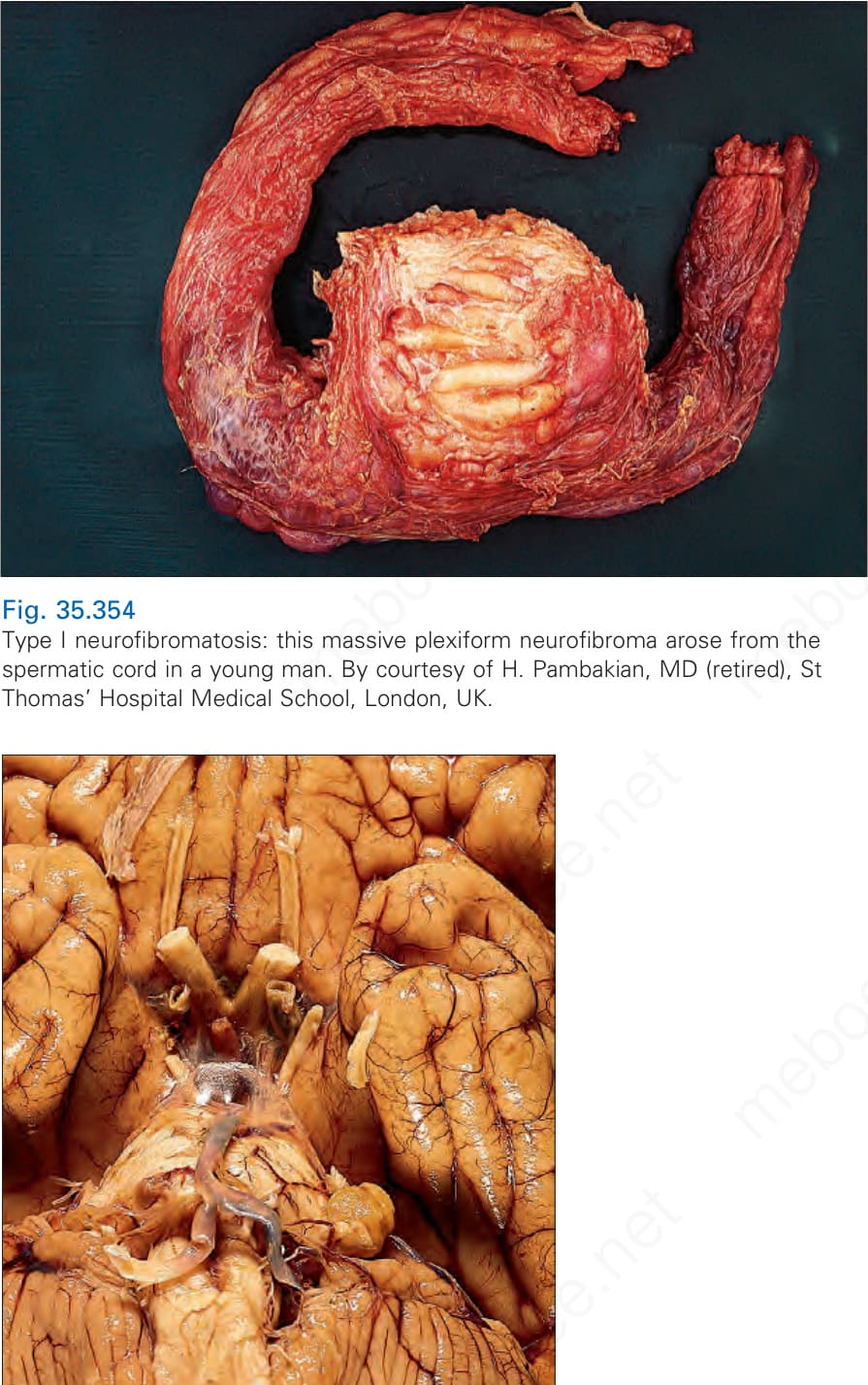
Fig. 35.354 Type I neurofibromatosis: this massive plexiform neurofibroma arose from the spermatic cord in a young man. By courtesy of H. Pambakian, MD (retired), St Thomas’ Hospital Medical School, London, UK.
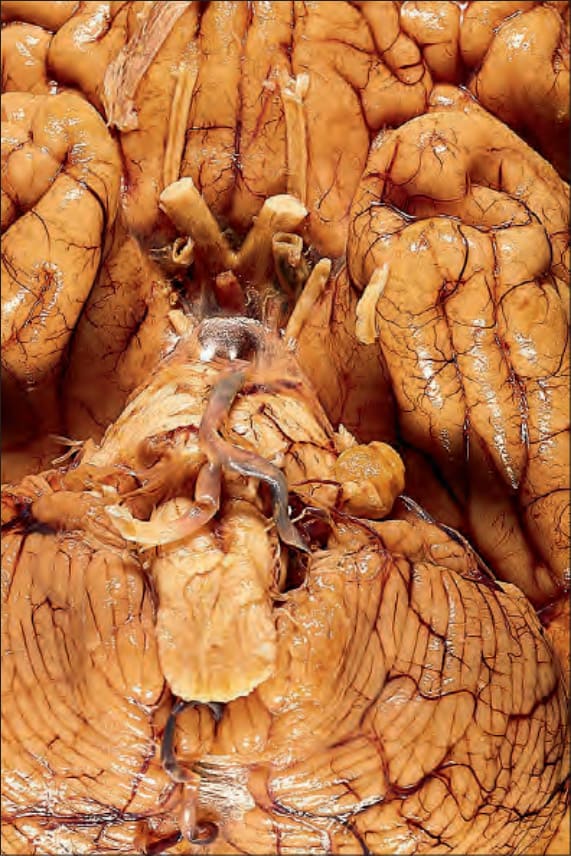
Fig. 35.355 Type I neurofibromatosis: a typical acoustic neuroma of the left eighth cranial nerve is visible in the cerebellopontine angle.