Histiocytic sarcoma
Histiocytic sarcoma
Histiocytic sarcoma (HS) (histiocytic lymphoma, malignant histiocytosis) is a very rare malignant proliferation of cells displaying morphological and immunophenotypic features of mature histiocytes.1,2 Many cases diagnosed before the era of immunohistochemistry and molecular techniques are now recognized to be examples of diffuse large B-cell lymphoma, anaplastic large cell lymphoma, or other NHLs.3–8 The historical literature is therefore difficult to interpret. However, there are a few well-documented, bona fide reports of HS including a few larger cohorts.2,8–23
Most patients are adults in the fourth or fifth decade of life but the age range is wide.2,8,19,23 A male predominance is reported in some series.8,19,23 The majority of lesions appear to arise at extranodal sites, particularly the intestinal tract, skin, and soft tissues.1,2,8,9,15,16,18–23 Involvement of the spleen, CNS, and other extranodal sites is rare.10–13,17,21,23,24 Cutaneous lesions are most often described as red or violaceous nodules. Rare cases present with systemic disease and multiple sites of involvement (‘malignant histiocytosis’).7,23,25 Some patients present with fever, weight loss, hepatosplenomegaly, and pancytopenia.8,19,23
In IDC sarcoma, tumor cells are consistently S100 and vimentin positive, and CD1a and langerin negative. Variable, often weak, reactivity is seen with antibodies to CD68, lysosyme, and CD45. FDC markers (CD21, CD23, CD35, CAN.42), myeloperoxidase, CD34, markers of B- and T-cell lineage, CD30, EMA, and cytokeratins are negative. The background small lymphocytes are almost always T cells.6,7,9,19
The majority of patients (70%) present with advanced-stage disease (stage III or IV), which is associated with a 60–80% mortality.1,2,8,23 However, more localized disease may be seen, in which case the outcome appears more favorable.19,23
Pathogenesis and histologic features The etiology and pathogenesis of HS are unknown. Few cases are associated with mediastinal germ cell tumors, mostly malignant teratoma plus or minus yolk sac tumor. This raises the possibility that HS derives from the same progenitor cell as the germ cell tumor.26,27 Other cases occur in
1511 Histiocytic sarcoma
association with other hematological malignancies, including NHL, myelodysplasia, and leukemia.2,8,19,23,28 In a small cohort of tumors, the same BCL2 translocation and/or immunoglobulin gene rearrangement as the follicular lymphoma with which they were associated was demonstrated.29 Thus, some neoplasms may arise as a consequence of transdifferentiation, alterations in transcription factor expression resulting in malignant lymphoid cells being diverted to a histiocytic pathway of differentiation.29 Mutations of the BRAF gene have been identified in a relatively high proportion of cases in one study.30
Histologically, there is a diffuse, often ‘bottom heavy’, extension from the reticular dermis into the subcutis (Figs 29.336 and 29.337). Epidermotropism is not a feature and a grenz zone is frequently evident.23 Tumor cells are large with moderate to abundant eosinophilic cytoplasm and often with fine vacuoles (Fig. 29.338). Nuclei tend to be eccentrically placed. They range from round to oval vesicular to multilobed, twisted, and sometimes bizarre variants occasionally mimicking Hodgkin or Reed-Sternberg cells.3,14,20 Nucleoli are prominent and frequently multiple. Mitotic activity is variable; mitoses are generally conspicuous and often abnormal.8,19,23 Spindle cell sarcoma-like features with a storiform growth pattern and xanthomatous forms may occur.19,20,23 Erythrophagocytosis is infrequent.23 Lymphophagocytosis may be a feature.19,20,23 Areas of necrosis may be seen, but are not usually extensive, and vascular invasion is rare.8,19,22 A prominent inflammatory cell infiltrate consisting of lymphocytes, neutrophils, and occasionally eosinophils and plasma cells is sometimes seen.1,19,23
myeloperoxidase), melanocytic (HMB-45, Melan A), and epithelial (cytokeratins) are absent.1,8,19,23
Ultrastructurally, the tumor cells have conspicuous lysosomes. Birbeck granules, desmosomal attachments, and interdigitating junctions are absent.8,14,23
Immunohistochemistry shows positivity for CD163, CD68, and/or lysosyme, the latter often showing accentuation in the Golgi region (Figs 29.339 and 29.340).1,8,19,23,31 There is often weak positive staining for CD45, CD45RO, and CD4, and occasionally weak expression of CD15.1,19,21–23 Staining for S100, usually weak and focal, is sometimes seen, suggesting a degree of dendritic cell differentiation.8,19,21,23 CD1a, and even langerin staining, is exceptional but only in a very small minority of cells.19,32 In such instances, it may be difficult or impossible to distinguish HS from LCS. Markers of other specific lines of differentiation, including FDC (CD21, CD35), lymphocyte (CD3, CD5, CD20, CD79a, pax5), myeloid (CD33,
In most cases, the antigen receptor genes are in a germline configuration. However, clonal immunoglobulin and T-cell receptor gene rearrangements have been rarely documented.3,28 The explanation for this may be transdifferentiation from pre-existing NHLs, or the so-called ‘lineage promiscuity’ due to the primitive nature of the neoplastic cells.2,8,29
Differential diagnosis Diagnosis of HS is dependent on demonstrating a histiocytic lineage in the tumor cells, while excluding other lines of differentiation, and is often a diagnosis of exclusion. Undifferentiated diffuse large B-cell lymphoma, anaplastic large cell lymphoma, and other NHLs can usually be recognized or reliably excluded if a broad immunohistochemical panel is employed. Melanocytic neoplasms usually express HMB-45 and/or melan A, and undifferentiated carcinomas one of the many keratins. Acute myeloid leukemia (AML) should react with CD33 and sometimes myeloperoxidase.
1512 Cutaneous lymphoproliferative diseases and related disorders
CUTANEOUS LEUKEMIC INFILTRATES AND PRECURSOR CELL NEOPLASMS
Introduction
Leukemias are neoplastic proliferations of leukocytes and their precursors in the bone marrow and peripheral blood. The subtypes can be broadly categorized into those of myeloid and lymphoid cells, as well as into tumors of mature leukocytes (‘chronic leukemias’) or their blast-cell precursors (‘acute leukemias). Not surprisingly, in view of high levels of circulating tumor cells, leukemic infiltrates often involve other tissues. The manifestations of several of the chronic leukemias (e.g., CLL, adult T-cell lymphoma/leukemia) are discussed elsewhere in this chapter, as are several lymphomas and myeloproliferative neoplasms that may present in skin and have a leukemic phase (e.g., MCL, mastocytosis). Chronic myeloid and myelomonocytic leukemias may also involve the skin, but only rarely.1–3 This section deals primarily with malignant neoplasms of hematopoietic precursor cells that may present with cutaneous disease, i.e., AML, blastic plasmacytoid dendritic cell neoplasm, and precursor lymphoblastic leukemia/lymphoma (LLL).
plasmacytoid dendritic cell neoplasm and MS remain as distinct entities, the latter as a unique presentation of AML of any subtype.12
Tumors of lymphoid precursors are referred to as lymphoblastic leukemias and/or lymphomas. They are subdivided into those of T-cell type and those derived from B cells. B-LLL is further subclassified according to the presence of specific genetic abnormalities that correlate with distinctive clinical or phenotypic properties with prognostic implications.13–15 As with AML, diagnostic criteria have been refined and new provisional entities added to both T- and B-cell categories.12
A detailed account of precursor cell neoplasms is beyond the scope of this textbook, and the following is only a very general overview. Historically, the French–American–British (FAB) system was used to classify cases of AML. This was morphologically based on and related to the type and degree of differentiation of the constituent blast cells. However, over recent years it has become increasingly apparent that a number of specific genetic alterations are found in AML, and these predict for clinical behavior and outcome. These were incorporated into the 2001 WHO classification, and used to define a category of ‘AML with recurrent genetic abnormalities’. Other categories recognized at this time included AML with multilineage dysplasia, therapy-related AML, and AML, not otherwise categorized. The latter group was subdivided on morphological grounds using a modified FAB approach, and included cases of myeloid sarcoma (MS).4–9
The dermatological manifestations of leukemia include a wide variety of non-specific lesions such as purpura and ecchymoses, adverse drug reactions, opportunistic infections, acute or chronic graft-versus-host disease, leukocytoclastic vasculitis, pyoderma gangrenosum, Sweet disease, and a rare intraepidermal vesicular eruption mimicking transient acantholytic dermatosis (Grover disease) and Hailey-Hailey disease.16–18 Leukemic vasculitis may also be encountered.19 Non-specific cutaneous lesions may also be seen in patients with myelodysplastic syndromes, hematological disorders that are characterized by chronic refractory cytopenias, infections, and hemorrhage.2,3,20,21 These include cutaneous vasculitis, photosensitivity, prurigo nodularis, Sweet disease, pyoderma gangrenosum, erythema elevatum diutinum, subcorneal pustular dermatosis, and relapsing polychondritis.22–28
The updated WHO classification published in 2008 further refined this classification. Two changes are of particular relevance to the skin. First, MS was recognized as a specific subtype for the first time. In addition, the entity previously known as ‘blastic NK cell lymphoma’ was renamed ‘blastic plasmacytoid dendritic cell neoplasm’, and moved from the general category of neoplasms of mature T cells and NK cells into the category of variants of AML.10,11 Although the principles of classification remain the same, the 2016 update of the WHO classification further refine the diagnostic criteria and categories to accommodate advances in knowledge, particularly with respect to recent discoveries in gene mutations.12 Importantly, blastic
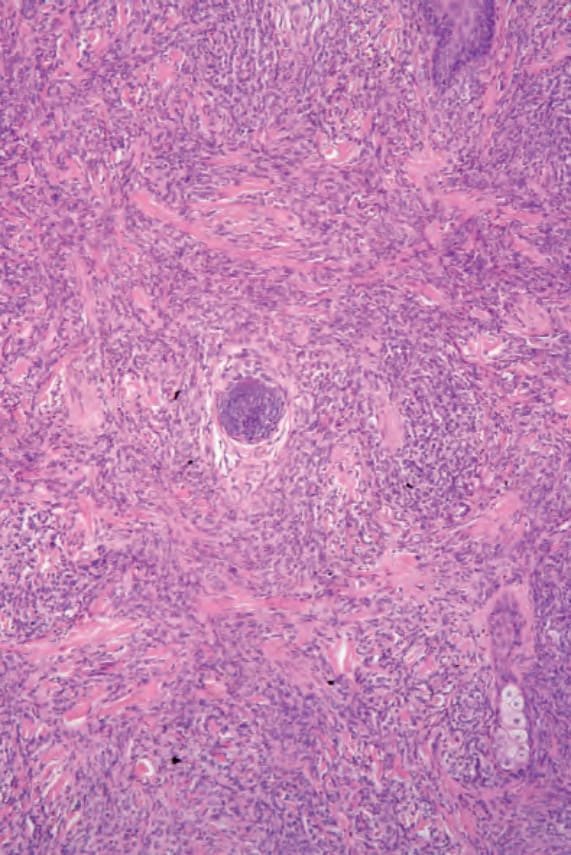
Fig. 29.336 Histiocytic sarcoma: there is a dense cellular infiltrate extending from the dermis into subcutaneous fat. By courtesy of C.D.M. Fletcher, MD, Brigham and Women’s Hospital and Harvard Medical School, Boston, USA.
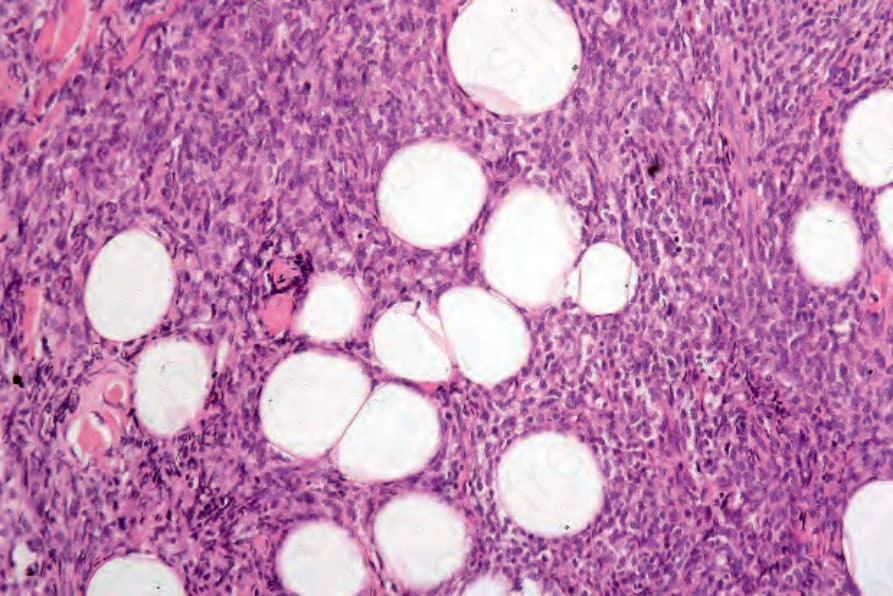
Fig. 29.337 Histiocytic sarcoma: the infiltrate extends into the subcutaneous fat. By courtesy of C.D.M. Fletcher, MD, Brigham and Women’s Hospital and Harvard Medical School, Boston, USA.
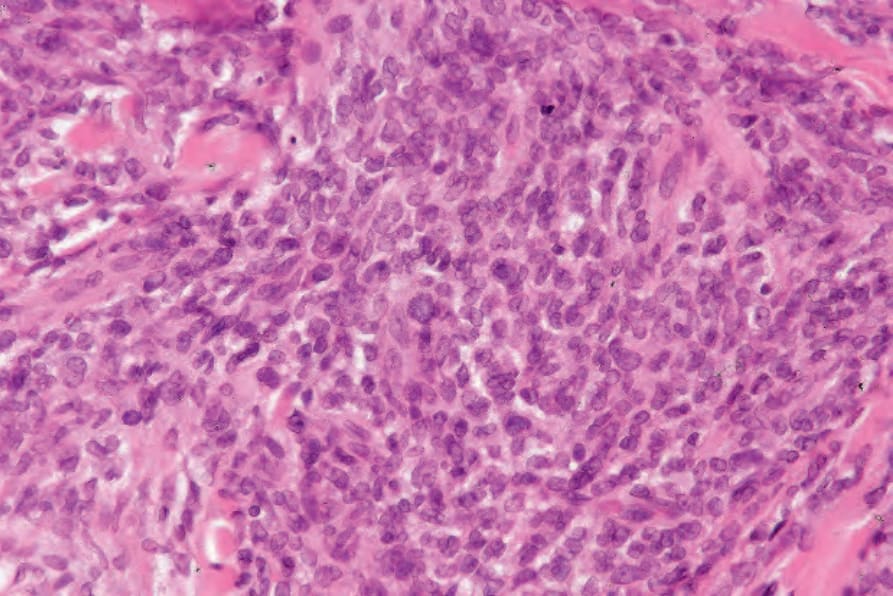
Fig. 29.338 Histiocytic sarcoma: the histiocytes have eosinophilic cytoplasm and vesicular nuclei. By courtesy of C.D.M. Fletcher, MD, Brigham and Women’s Hospital and Harvard Medical School, Boston, USA.
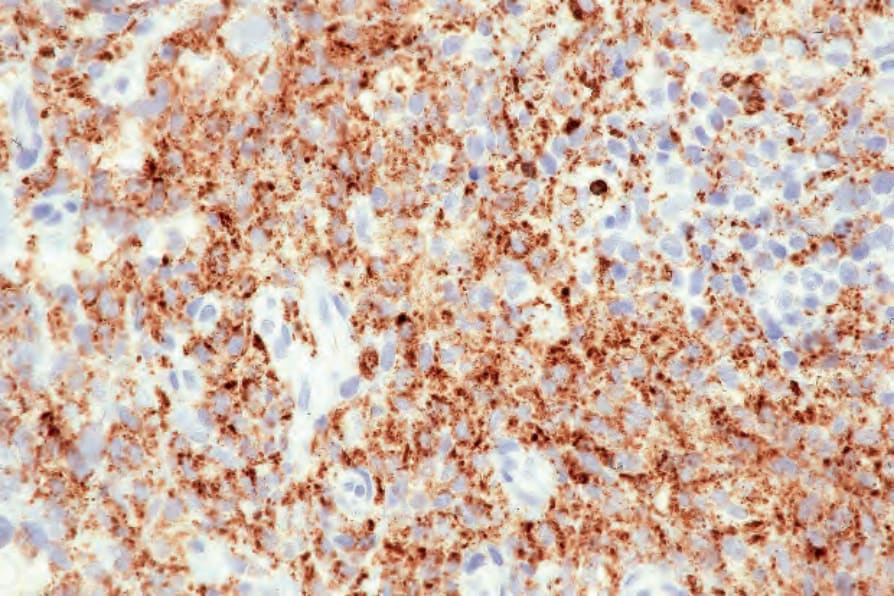
Fig. 29.339 Histiocytic sarcoma: the tumor cells express CD68. By courtesy of C.D.M. Fletcher, Brigham and Women’s Hospital and Harvard Medical School, Boston, USA.
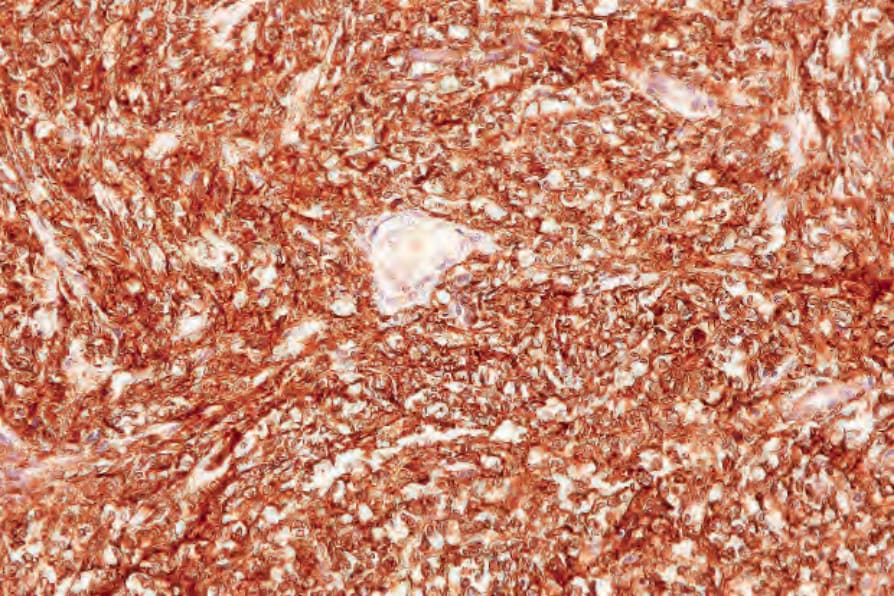
Fig. 29.340 Histiocytic sarcoma: note the diffuse lysozyme expression. By courtesy of C.D.M. Fletcher, Brigham and Women’s Hospital and Harvard Medical School, Boston, USA.