Crystal-storing histiocytosis
Crystal-storing histiocytosis
Clinical features Crystal-storing histiocytosis is an extremely rare condition characterized by deposition of crystalline material in the cytoplasm of histiocytes.1–3 About 90% of the cases have been described in patients with lymphoplasmacytic neoplasms, including lymphoplasmacytic lymphoma (immunocytoma), monoclonal gammopathy of uncertain significance, multiple myeloma, extramedullary plasmacytoma, MALT lymphoma, and large cell B-cell lymphoma.1,4–8 Much more infrequently (in up to 10% of the cases), crystal-storing histiocytosis has been associated with non-neoplastic disorders, including various inflammatory diseases (e.g., rheumatoid arthritis, pulmonary infections, and Crohn disease), metabolic states (cystinosis), and drugs (clofazimine).1 Localized variant, defined as a single deposit involving only one organ or site, and generalized variant of the disease, involving two or more distant organs or sites, have been recognized.1
Familial HLH occurs in around 1 in 50 000 live-born children and usually presents at <1 year of age, and exceptionally in older children and adults.4–6 Acquired HLH tends to occur in older age groups.
The diagnostic criteria for HLH have been revised and are based on the signs and symptoms typically seen in hemophagocytic syndromes.1 To establish the diagnosis, five of the following criteria should be present:
• fever,
• splenomegaly,
• cytopenias affecting more than two lineages,
• hypertriglyceridemia and/or hypofibrinogenemia,
• hemophagocytosis in bone marrow, spleen or lymph nodes,
• no evidence of malignancy,
• low or absent NK-cell activity,
• raised ferritin; and raised soluble CD25. Alternatively, primary forms of the disease may be diagnosed if a specific genetic mutation consistent with familial HLH can be identified.1
Primary cutaneous involvement is exceptional, and the literature on this aspect is limited to six cases only, all associated with lymphoplasmacytic neoplasms.3,9–11 These patients presented with either swelling, indurated plaques, pruritic rash, or erythematous asymptomatic subcutaneous nodules and tumors (Fig. 29.330) with no particular site of predilection. Cutaneous crystal-storing histiocytosis can develop early in the course of neoplastic disease and is not necessarily associated with an aggressive clinical course.3
Other clinical and laboratory findings often seen in HLH include cerebromeningeal symptoms, lymphadenopathy, jaundice, edema, skin rashes, abnormal liver function tests, hypoproteinemia, hyponatremia, raised very low density lipoprotein (VLDL), and reduced high density lipoprotein (HDL).1 Skin manifestations are seen in up to 65% of patients and include a transient purpuric or hemorrhagic maculopapular eruption, erythroderma, and morbilliform erythema.7
Histologic features The condition is characterized by the presence of histiocytes containing eosinophilic crystals which have been likened to Gaucher cells (pseudo-Gaucher cells) admixed with lymphoma cells (Fig. 29.331).6 The crystals are variably polygonal to rhomboid or needle-shaped and stain positively with PAS and phosphotungstic acid hematoxylin (Fig. 29.332).9,10 The crystals are usually composed of kappa light chain with no consistent association with any particular heavy chain.3 Erythrophagocytosis may also be evident.6
Despite recent advances in treatment, only about 50% of patients are cured.1,3,8
Pathogenesis and histologic features Natural killer (NK) cells play an important role in regulating the macrophage response following activation by various infectious agents. In all types of HLH, there appears to be a defect in NK target cell killing leading to uncontrolled activation of macrophages.1,3,9,10 Familial HLH is characterized by defects in genes critical to cytotoxic granule formation, movement, or exocytosis. About 15–50% of cases have mutations in the perforin gene, 15–30% defects in the UNC13D gene, and other mutations in the STX11 gene.3,11–19 Inherited disorders that predispose to the development of HLH also typically harbor defects in genes involved in cytotoxic granule formation and/or function, or key regulatory signaling effects of lymphocytes,
The histiocytes express CD68, and sometimes immunoglobulin heavy and light chains may be stained weakly.6
Ultrastructurally, the crystals are sometimes membrane bound (of lysosomal derivation) and display platelike, rectangular, trapezoid, and rhomboid shapes with a distinct hexagonal lattice structure.6,9,10 The tumor cells may also contain intracytoplasmic crystals.6
Follicular dendritic cell sarcoma and interdigitating dendritic cell sarcoma
Clinical features Follicular dendritic cell (FDC) sarcoma and interdigitating dendritic cell (IDC) sarcoma are exceptional neoplasms of antigen-presenting dendritic cells. FDCs are of mesenchymal origin, reside in the primary and secondary lymphoid follicles, and play a major role in the induction and maintenance
1509 Follicular dendritic cell sarcoma and interdigitating dendritic cell sarcoma Crystal-storing histiocytosis
intestine, mesentery, spleen, and testes, among other sites.3,6,11,12 Skin involvement in both types of tumor is very rare, and is often part of disseminated disease.3,6,13–20
Disease is localized at presentation in the majority of cases (77%), more frequently in FDC (85%) than IDC sarcoma (60%).8 Clinical behavior is more that of a low-grade soft tissue sarcoma than lymphoma, with an intermediate risk of local recurrence and distant metastasis (≈30%).3,6,21,22 Intra-abdominal location and large tumor size have also been associated with a poorer prognosis.21,23
Pathogenesis and histologic features An association between FDC sarcoma and hyaline vascular CD has been reported, and it has been proposed that FDC proliferation in CD predisposes to dysplastic change, with evolution into FDC sarcoma.3,17,21,24–26 EBV-encoded RNA is present in almost all tumor cells in a subset of IPT-like FDC sarcomas arising in the liver and spleen. The virus has been shown to be present in monoclonal and episomal forms.27–31 BRAF mutations have been identified in FDC sarcoma, as have loss of function alterations in tumor suppressor genes involved in negative regulation of NF-κB activation and cell cycle progression.32,33 In addition, some FDC sarcomas harbor copy number gains of 9p24, implicating PD-1L and PD-2L and immune evasion in disease pathogenesis.34 A proportion of IDC sarcoma are also associated with hematological malignancies including CLL, follicular lymphoma, T-lymphoblastic leukemia, diffuse large B-cell lymphoma, and mycosis fungoides.6
The neoplastic cells in FDC sarcoma are spindled to oval in shape with eosinophilic cytoplasm and indistinct cell borders. Nuclei are oval or elongated with vesicular or finely granular chromatin, and generally small but distinct nucleoli. (Figs 29.333–29.335).3,19,21 Nuclear pseudoinclusions are common and binucleate and multinucleate cells may be seen. Most cases appear cytologically bland, but significant atypia may be seen in some cases. The growth pattern is fascicular, storiform, sheetlike, or nodular. The mitotic count is variable (0–50/10 HPF) but usually relatively low (0–10/10 HPF). Areas of necrosis may be seen. Small lymphocytes, scattered between the tumor cells and forming perivascular aggregates, are common.3,7,8,11
of the humoral immune response.1,2,3 IDCs are bone marrow-derived cells, typically found in the T-cell areas of peripheral lymphoid organs, and they stimulate resting T cells during a primary immune response.2,4,5
FDC and IDC sarcomas occur in a wide age range, including children, but most patients are adults, mainly in the fifth decade.3,6–12 Sex distribution is about equal, apart from IPT-like FDC sarcomas, in which there is a marked female predominance.3,6,7–9,11 Both tumor types arise most commonly in lymph nodes, particularly cervical lymph nodes for FDC sarcoma. Up to one-third of cases present at extranodal sites.3,6,7–9 Extranodal FDC sarcoma has been most frequently reported in the upper aerodigestive tract (oral cavity and tonsil), soft tissues, liver, and gastrointestinal tract, whilst IDC sarcoma has been documented in the mediastinum, nasopharynx,
IDC sarcoma shows a similar fascicular, storiform, and/or whorled growth pattern, and involves the T-cell areas in nodal forms. The tumor cells are large fusiform spindle-shaped cells with abundant eosinophilic cytoplasm and indistinct cell borders. Nuclei are spindled to ovoid with finely dispersed chromatin and distinct nucleoli of small to large size. Occasional multinucleate cells may be seen and the degree of cytological atypia is variable. The mitotic count is usually low (<5/10 HPF) and necrosis is rare. Lymphocytes, and less commonly plasma cells, are usually present. The morphology is very similar to, and often indistinguishable from, FDC sarcoma.6,7,9
1510 Cutaneous lymphoproliferative diseases and related disorders
Ultrastructurally, cells in FDC sarcoma are characterized by villous processes united by desmosomal attachments.21 Birbeck granules and lysosomes are absent. IDC sarcoma possesses complex interdigitating cell junctions lacking well-formed desmosomes. Scattered lysosomes may be present but there are no Birbeck granules.19
Differential diagnosis FDC and IDC sarcomas are underrecognized, particularly at extranodal sites, and up to one-third of cases are misdiagnosed at initial evaluation.11,19,39 Once FDC and IDC sarcoma enter the differential diagnosis, appropriate immunohistochemistry should permit reliable distinction from each other, and from other neoplasms, such as Langerhans cell histiocytosis and LCS, HS, melanoma, and spindle cell carcinoma.
Tumor cells in FDC sarcoma react positively with one or more FDC makers, including CD21, CD23, CD35, KiMP4, and CAN.42.3,7,8,19 CXCL13 and clusterin have been proposed as useful markers for FDC sarcoma, particularly with respect to other dendritic cell tumors.34–36 Staining for vimentin, fascin, epidermal growth factor receptor, HLA-DR, and desmoplakin is often positive. CD68, S100, and EMA are variably expressed.7,19,37,38 Cytokeratin is very rare and when present is seen in <10% of neoplastic cells.11 Rarely, tumor cells express CD20 and/or CD45.19 Staining for CD1a, lysosyme, myeloperoxidase, CD34, CD3, CD79A, CD30, and HMB-45 is negative. The admixed small lymphocytes may be predominantly B or T cells or a mixture of both.19,21
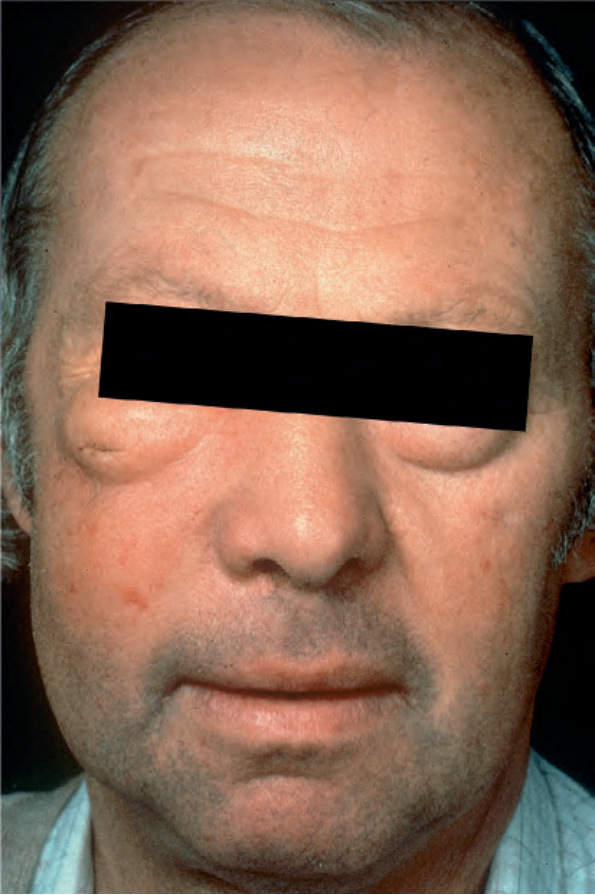
Fig. 29.330 Crystal-storing histiocytosis: this patient presented with marked swelling of the face and the eyelids.
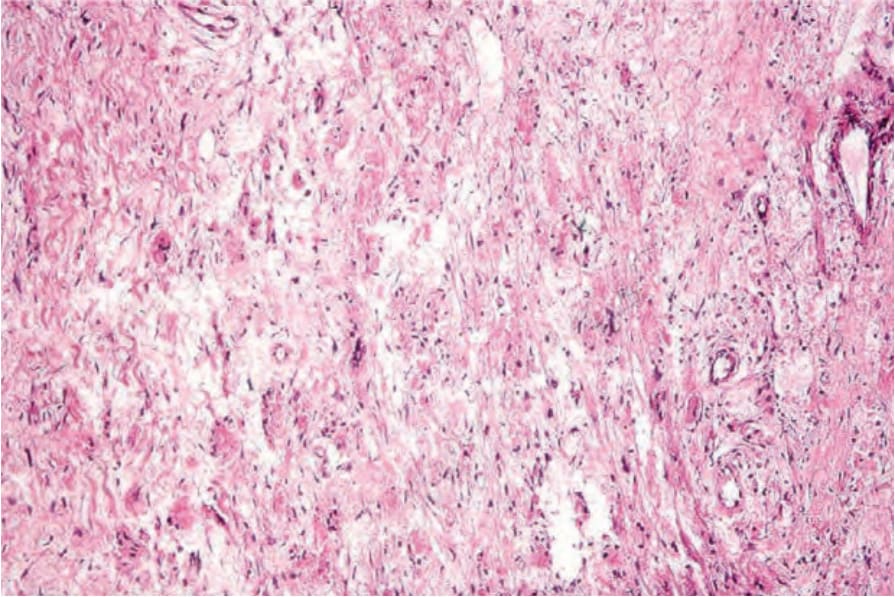
Fig. 29.331 Crystal-storing histiocytosis: within the dermis are large histiocytes with abundant eosinophilic cytoplasm.
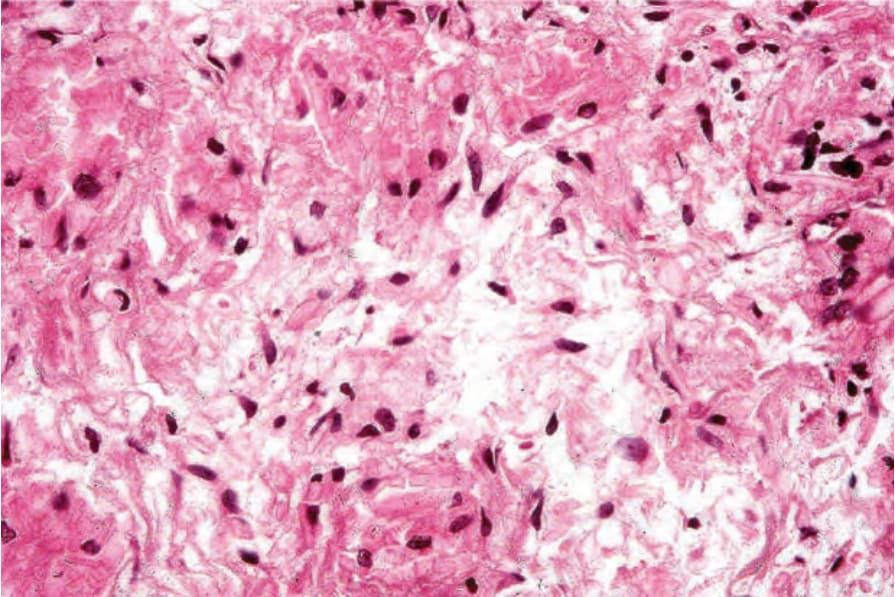
Fig. 29.332 Crystal-storing histiocytosis: the cytoplasm contains large eosinophilic crystals.
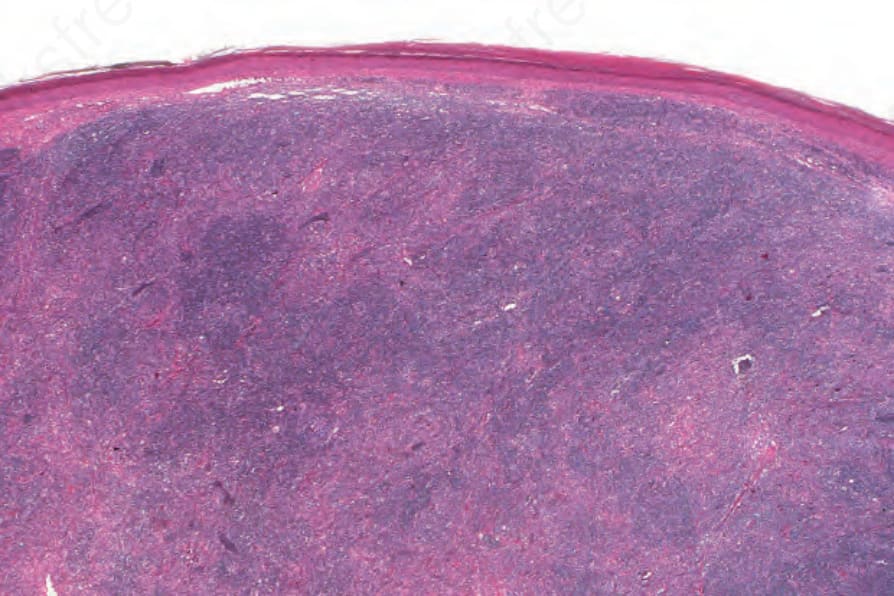
Fig. 29.333 Follicular dendritic cell tumor: there is a heavy dermal infiltrate consisting of pale-staining follicular dendritic cells associated with a background population of lymphocytes. By courtesy of N.L. Harris, MD, Massachusetts General Hospital and Harvard Medical School, Boston, USA.
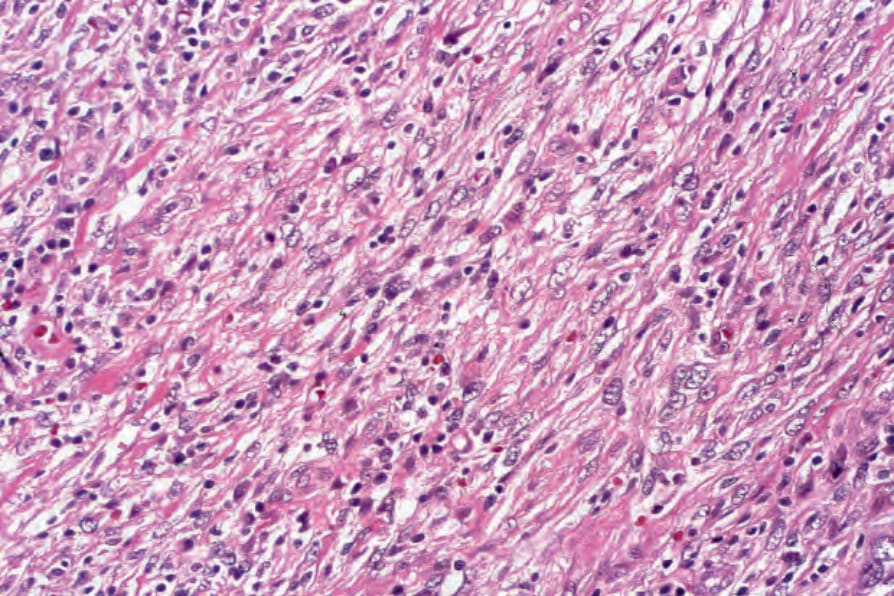
Fig. 29.334 Follicular dendritic cell tumor: the tumor cells are arranged in loose fascicles. By courtesy of N.L. Harris, MD, Massachusetts General Hospital and Harvard Medical School, Boston, USA.
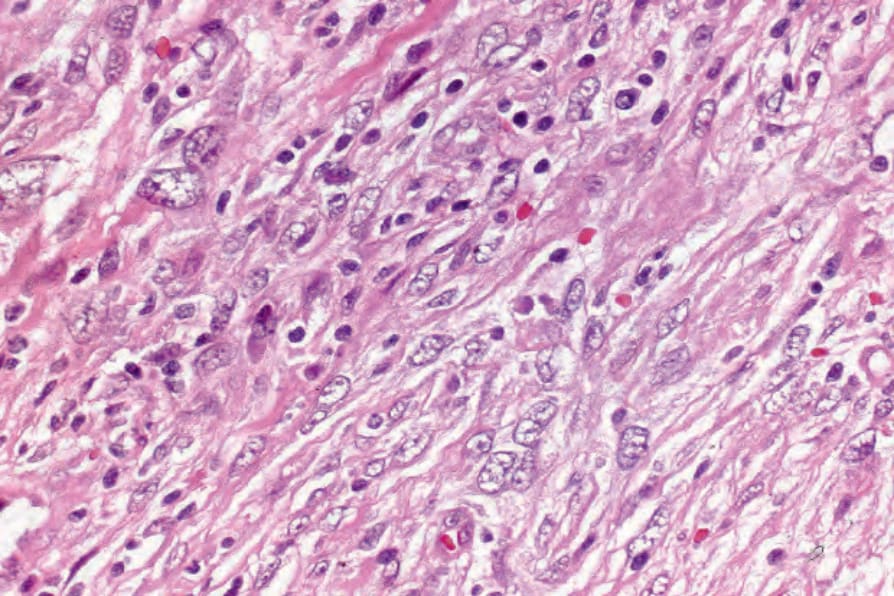
Fig. 29.335 Follicular dendritic cell tumor: the spindle cells have eosinophilic cytoplasm and elongated vesicular nuclei. By courtesy of N.L. Harris, MD, Massachusetts General Hospital and Harvard Medical School, Boston, USA.