Hemophagocytic lymphohistiocytosis
Hemophagocytic lymphohistiocytosis
Clinical features Hemophagocytic lymphohistiocytosis (HLH) (hemophagocytosis syndrome) represents a complex group of disorders that share a final common pathway culminating in hemophagocytosis and associated signs and symptoms. They are divided into primary and secondary HLH. Primary HLH equates with familial forms of the disease and is usually associated with an inherited genetic disorder, although a family history may be negative, and HLH in these patients may be triggered by infections.1,2 Secondary variants include
1508 Cutaneous lymphoproliferative diseases and related disorders
e.g., mutations in LYST in Chediak-Higashi syndrome or SH2D1a, or SAP in X-linked lymphoproliferative syndrome.3,20–22 Molecular defects predisposing patients to other secondary HLH disorders have yet to be delineated.
The levels of many cytokines are raised in HLH, mostly as a consequence of this loss in regulatory processes, and they may account for some of the typical signs and symptoms. For example, raised interleukin-1 may be a cause of fever, and increased levels of TNF-α and IFN-γ may account for fatigue, wasting, and pancytopenia. TNF-α has also been implicated in down-regulation of lipoprotein lipase, explaining the raised serum triglycerides.3
HLH associated with inherited immune deficiencies (e.g., Chédiak-Higashi syndrome, X-linked lymphoproliferative syndrome, and Griscelli syndrome), and acquired HLH in patients with immune system defects. In the latter, HLH is triggered by severe infection with viruses (especially EBV and CMV), other infectious agents (e.g., leishmaniasis) or malignancy (particularly various lymphomas). HLH may also complicate autoimmune disease, including macrophage activation syndrome in systemic juvenile arthritis, adult-onset Still disease, and lupus erythematosus.3
Histologically, the infiltrate consists of mature lymphocytes (including activated forms) and macrophages showing phagocytosis of any or all hemopoietic elements, and particularly affecting the spleen, lymph nodes, bone marrow, liver, and CSF.23 Cutaneous involvement occurs in less than 10% of cases, and the features are usually non-specific, consisting of a perivascular lymphohistiocytic infiltrate. Hemophagocytosis is rarely a feature.7,23 The histiocytes are CD68 positive.
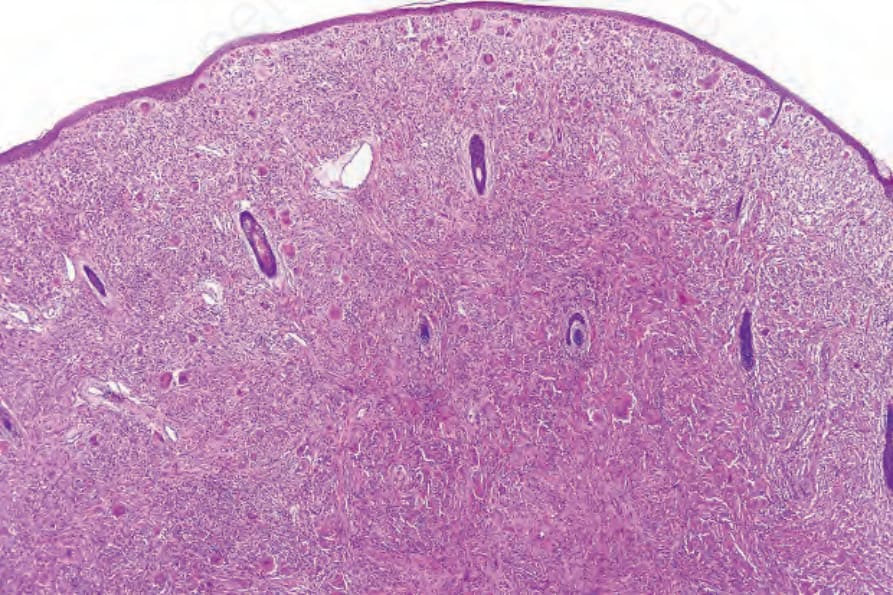
Fig. 29.325 Reticulohistiocytoma: the dermis is expanded by a dome-shaped nodule composed of large histiocytes.
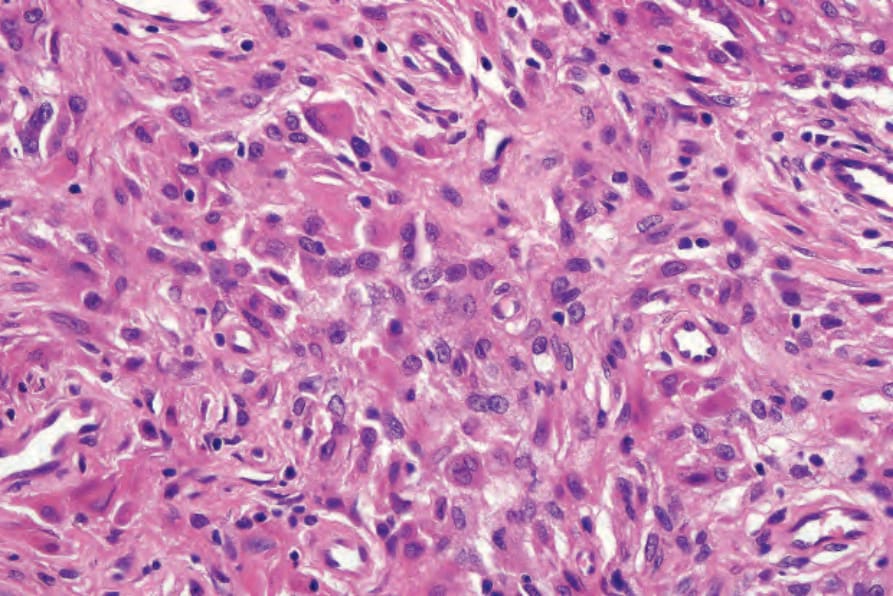
Fig. 29.326 Reticulohistiocytoma: the histiocytes have copious eosinophilic cytoplasm.
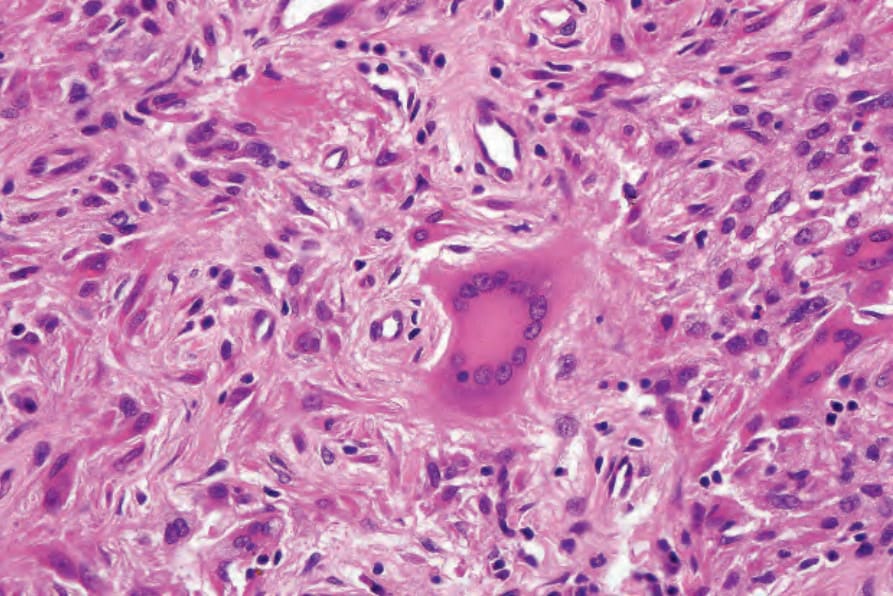
Fig. 29.327 Reticulohistiocytoma: characteristic multinucleate giant cell with ground-glass cytoplasm.
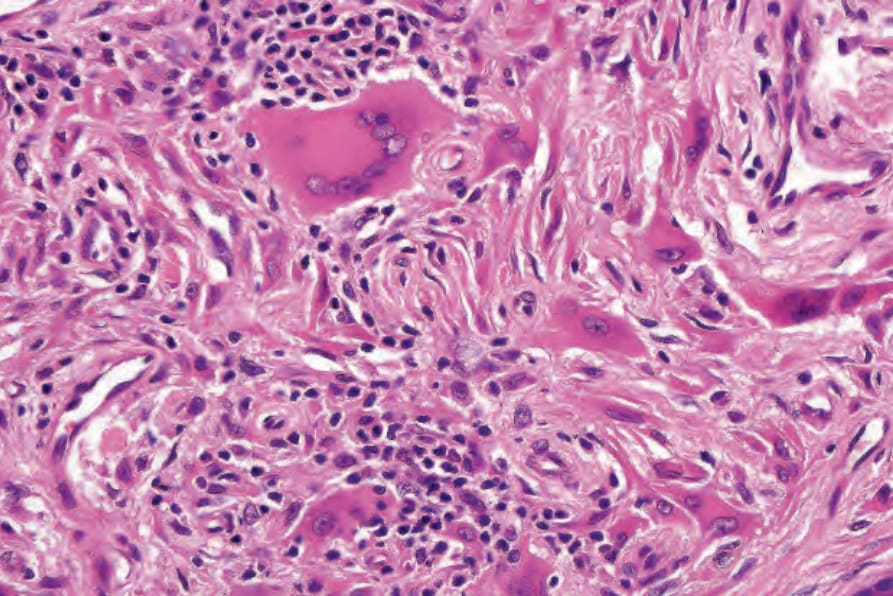
Fig. 29.328 Reticulohistiocytoma: lymphocytes are also present.
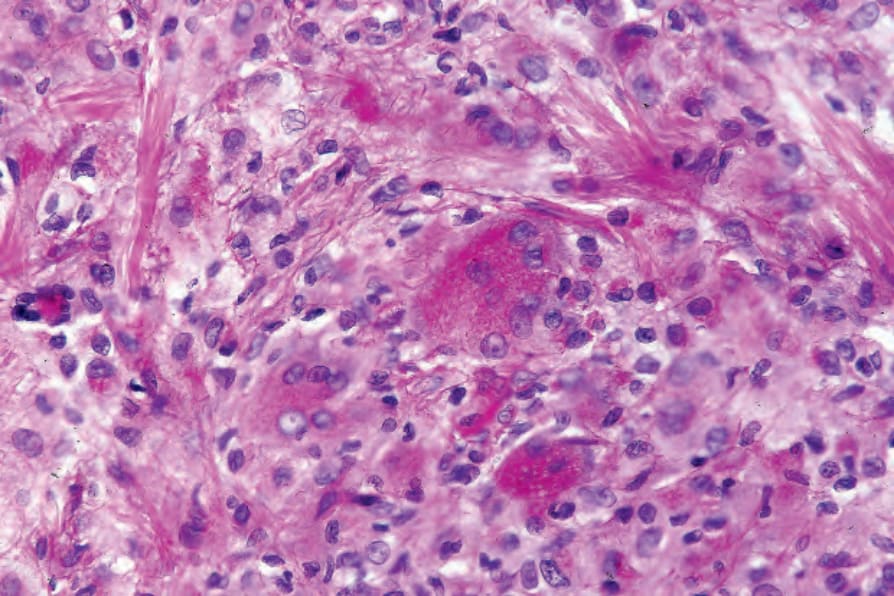
Fig. 29.329 Reticulohistiocytoma: the giant cells are PAS positive, diastase resistant.