Rosai-Dorfman disease
Rosai-Dorfman disease
Clinical features Rosai-Dorfman disease (RDD) (sinus histiocytosis with massive lymphadenopathy) is a rare, benign, self-limiting illness characterized by a reactive
1503 Rosai-Dorfman disease
A
proliferation of histiocytes that phenotypically appear to be macrophage– dendritic cell hybrids.1–3 Age range is wide with predilection for the first and second decades of life and a slight male predominance.1–3 Blacks and whites are most often affected, while it is rare in Asians.4 Clinically, it is characterized by massive, painless, frequently bilateral, cervical lymphadenopathy associated with pyrexia, night sweats, leukocytosis with neutrophilia, raised ESR, and a polyclonal hyperglobulinemia.1–5 Occasionally, other groups of lymph nodes may be involved. Extranodal deposits are common (43% of cases) and are most frequently encountered in skin and subcutis, orbit, bone, and CNS coverings, although virtually any organ can be affected.2,4–10
Skin lesions are present in 9% of cases and are not uncommonly seen in the absence of lymph node and other extracutaneous disease, or systemic abnormalities.5–8,11–17 The latter situation, referred to as cutaneous RDD, seems to occur in a somewhat older age group (fifth decade of life; range,
B
1504 Cutaneous lymphoproliferative diseases and related disorders
15–77 years), is more frequent in females, and has a higher prevalence in Asians compared to classic nodal disease.11,13,16–18
Cutaneous disease presents as xanthomatous, erythematous, or red-brown papules, nodules, or plaques, that may be single, clustered, or widespread (Figs 29.315–29.318). The trunk and limbs are most frequently involved.7,13–17 Acne rosacea-like, giant granuloma annulare-like, acneiform, and pustular lesions, and a vasculitis-like appearance have rarely been described.12,14,19,20 Subcutaneous lesions also occur.21
Most patients with RDD run a benign clinical course with partial or complete spontaneous remission, although this may take a number of years, and episodes of relapse are possible.2,4,9 Residual atrophic brown macules may persist after resolution.3 Fatalities are encountered in approximately
5% of patients, as a result of related immunological abnormalities or malignancies (including HL, NHL, leukemia, or solid tumors), or due to the secondary effects of disease at vulnerable sites such as the CNS or upper respiratory tract.3,9,22,23
Pathogenesis and histologic features The etiology is unknown. The proliferating histiocytes are polyclonal, and it has been postulated that RDD may represent a disorder of cell-mediated immunity due to cytokine dysregulation and/or an exuberant response to an infective (possibly viral) agent.14,19,24,25 There is no convincing evidence for an EBV infection.26 The role of human herpesvirus (HHV) is uncertain. Current evidence suggests that neither HHV6 nor HHV8 is implicated in the
1505 Rosai-Dorfman disease
majority of cases.27 Rosai-Dorfman-like changes are seen in association with other conditions. These include autoimmune hemolytic anemia, systemic lupus erythematosus, and juvenile idiopathic arthritis.28 Rosai-Dorfmanlike changes are also seen in 41% of patients with autoimmune lymphoproliferative syndrome type 1a who have mutations in TNFRSF6 affecting the FAS gene, as well as in patients in Faisalabad Histiocytosis and ‘H’ syndrome.29–31 Because of these varied associations, it may be that RDD represents a reaction pattern rather than a single distinct entity. BRAF mutations are not found.32,33
The lymph node findings are characteristic.2 The node sinuses are dilated and contain very large numbers of histiocytes, characterized by abundant, pale-staining eosinophilic cytoplasm with irregular (feathery) borders, large, centrally placed vesicular nuclei with a single distinct nucleolus.1,2,4 Mitotic figures are rare. Characteristically, the cytoplasm of the histiocytes appears to contain lymphocytes, red blood cells, and/or granulocytes, a feature known as emperipolesis. Some cells have foamy cytoplasm and some may be multinucleate. Plasma cells are common.
The skin lesions are characterized by a dense cellular infiltrate involving the dermis and sometimes the subcutis (Fig. 29.319).7,11,13,16,17 There may be associated acanthosis or, rarely, collarette formation. Ulceration is uncommon. The infiltrate typically has a nodular appearance, and occasionally a lobular pattern is seen (Fig. 29.320). It is composed of sheets of histiocytes similar to those described in the lymph node, although emperipolesis may be less conspicuous (Figs 29.321 and 29.322). Cytological atypia is absent and xanthomatized cells are rare. There is often an admixture of lymphocytes, plasma cells, neutrophils, and eosinophils (Fig. 29.323). An increase in vascularity is sometimes a feature, and in certain cases stromal fibrosis with a storiform pattern is seen.13,18 Rarely, lymphoid follicles with germinal centers are apparent.14,16 Intravascular involvement has been described.27
In rare cases, localized proliferations of Langerhans cells resembling microscopic foci of Langerhans cell histiocytosis are noted.16,34 It is not clear whether this represents transdifferentiation, coincidence of two separate pathologies, or a reactive Langerhans cell proliferation.
1506 Cutaneous lymphoproliferative diseases and related disorders
Multicentric reticulohistiocytosis and reticulohistiocytoma
Clinical features Multicentric reticulohistiocytosis (MRH) (lipoid dermatoarthritis, giant cell reticulohistiocytosis) is a rare non-Langerhans cell histiocytosis characterized by a cutaneous eruption, usually associated with a severe arthropathy, frequent mucous membrane involvement, and occasional visceral symptoms.1–3 Most patients are adults aged >40 years (mean age at presentation, 43 years; range, 6–71 years).3–7 There is female predilection (3 : 1).2–5 Two-thirds of patients present with arthropathy and invariably develop skin lesions within months to a few years. Cutaneous lesions represent the primary complaint in 20% of patients, and while many go on to develop joint symptoms, in some the disease remains in the skin (referred as generalized cutaneous reticulohistiocytosis).1,2 Cutaneous lesions present on the face (predominantly ears, nose, and paranasal areas), the hands (dorsum and lateral aspects of the fingers, nail folds), neck, and trunk. Multiple reddish-brown to yellow papules and nodules measuring from a few millimeters to 2 cm in diameter are seen.2–4,8–11 Sometimes they coalesce to form plaques with a cobblestone appearance. Nail fold changes present a characteristic ‘coral band’ pattern.5 The interphalangeal joints of the hand are the most frequently affected joints, and there may be shortening of the fingers with an ‘opera glass’ deformity when both proximal and distal interphalangeal joints are involved – so-called ‘main en lorgnette’.5,8 Less often, there is involvement of the knees, wrists, shoulders, hips, ankles, feet, and elbows, in decreasing order of frequency.2
The histiocytes express S100 protein, the macrophage markers CD68, CD14, and CD163 (both strong surface staining) (Fig. 29.324). Factor XIIIa, CD1a, and langerin are negative.
Ultrastructurally, the histiocytes have undulating villous cytoplasmic processes.1
Mucous membrane lesions are present in 50% of patients, and affect the oral cavity, pharynx, and nose. Involvement of other sites is rare and include the lungs, heart, liver, breast, stomach, thyroid, submandibular gland, skeletal muscle, and bone.2,12–22 Hyperlipidemia is reported in 30–50% of cases, and patients often have xanthelasmata.23 An internal malignancy is found in 30–50% of patients, including carcinoma of the breast and stomach and, more rarely, carcinoma of the cervix, colon, lung, and ovary, lymphoma, leukemia, sarcoma, and melanoma.23–32 An association with autoimmune disease is described in 15–27% of cases, including polymyositis, Sjögren syndrome, hypothyroidism, primary biliary cirrhosis, vitiligo, Hashimoto thyroiditis, and lupus erythematosus.1,3,5,33–35 Concomitant vasculitis is rarely seen.8 Some cases have been linked to infection.3 Non-specific symptoms including weight loss, fever, and weakness are rare.
Differential diagnosis Skin deposits in RDD may be confused histologically with eruptive xanthomata. However, histiocytes in xanthomata are usually S100 negative and lack emperipolesis. Langerhans cell histiocytosis can be differentiated by the smaller size of the histiocytes, the typical folded reniform nucleus, epidermotropism, and positive staining for CD1a and langerin.7 In reticulohistiocytosis, the histiocytes have prominent eosinophilic ground-glass cytoplasm and are usually negative for S100. In the JXG family of disorders, the histiocytes are often factor XIIIA positive, and negative or focally positive for S100. Some of the plasma cells in RDD disease may express IgG4, and some authors proposed that there is some overlap between RDD and IgG4-RD.35,36 Separation of these two entities should be based on recently published criteria that take into account pathological and clinical features as well as the serum IgG4 level.
The outcome of the disease is unpredictable. Cutaneous lesions may resolve spontaneously or persist. In around 50% of patients, joint symptoms remain stable or diminish over many years. However, in the remainder, disease is progressive with disabling and destructive arthropathy.4,8
RH (solitary epithelioid histiocytoma) refers to solitary cutaneous lesions, histologically identical to those in MRH.36–38 Patients are usually young adults (mean and median age at presentation, 35 years), although age range is wide. There is no sex predilection. Clinically, a papule or nodule measuring up to 1 cm in diameter on the skin or mucous membranes is seen. Trunk, head and neck, or extremities may be the presenting site but, unlike MRH, fingers are not involved.36
Pathogenesis and histologic features The pathogenesis of MRH and RH is unknown. An abnormal macrophage response to different stimuli has been proposed. An immunological basis for the histiocytic reaction in MRH is possible in view of the relationship with infection, autoimmune disease, and internal malignancy.9 There may be a more localized trigger in RH, but this has not been proven.39
1507 Hemophagocytic lymphohistiocytosis
and factor XIIIa. They are negative for CD1a and S100.1,2,36 The giant cells are diastase-PAS positive (Fig. 29.329).
Synovial lesions show similar histology.6
Differential diagnosis In cases with systemic features, the clinical history is paramount. Solitary RH may be distinguished from JXG by the absence of lipidized cells and Touton giant cells and the typical eosinophilic cells with ground-glass cytoplasm. However, JXG may have scattered cells similar to the latter. Distinction from a melanocytic lesion is made by negative staining for melanocytic markers.
Cutaneous lesions in MRH and RH are identical and consist of a well-defined collection of uniform pink histiocytes and multinucleated giant cells, with somewhat eosinophilic, finely granular, ground-glass cytoplasm. The infiltrate is mainly dermal, but rarely erodes the basal epidermis or extends into superficial subcutis (Figs 29.325–29.328). Scattered inflammatory cells, including lymphocytes, granulocytes, and plasma cells, may also be present.
The histiocytes and multinucleate giant cells are consistently positive for CD68, CD163, and Ki-M1p and react variably with antibodies to Ham56
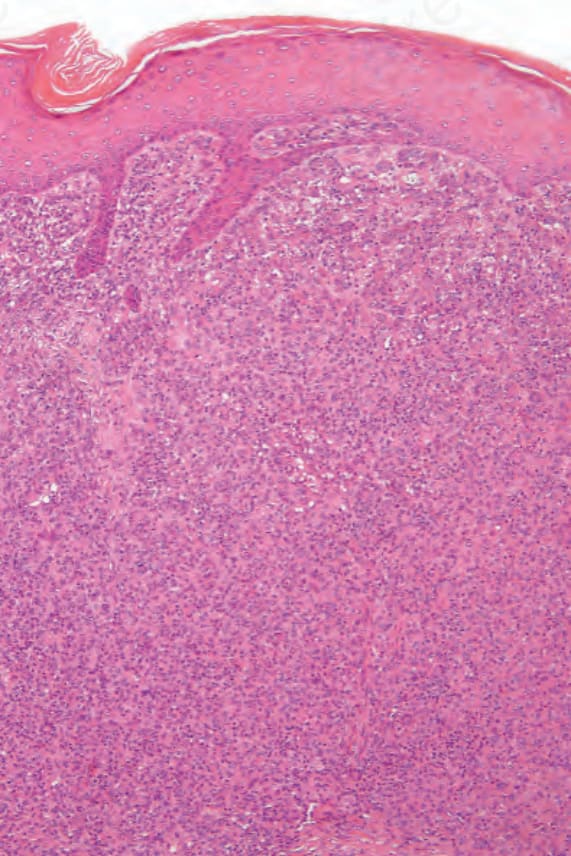
Fig. 29.310 Xanthogranuloma: medium-power view of Fig. 29.309.
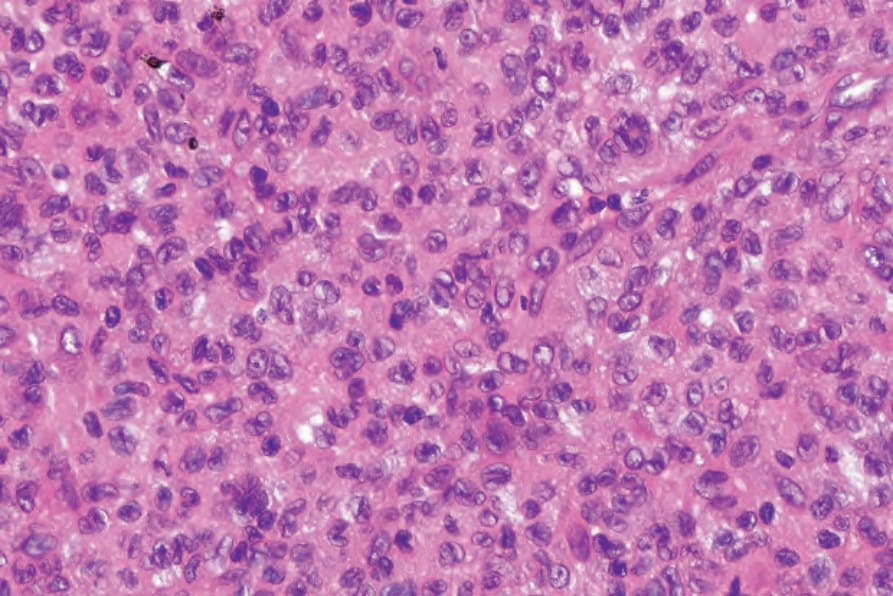
Fig. 29.311 Xanthogranuloma: in early lesions xanthomatized forms are often absent.
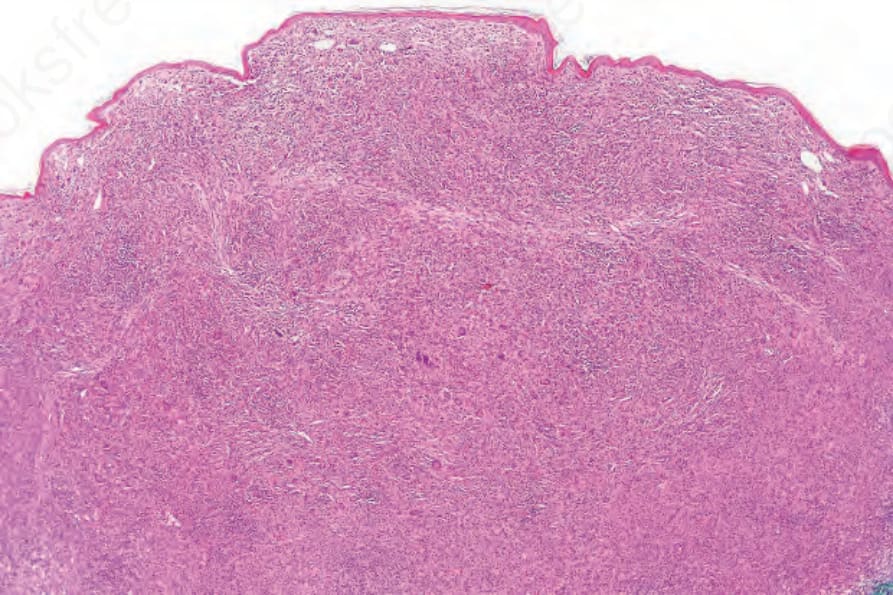
Fig. 29.312 Xanthogranuloma: scanning view of a dermal nodule.
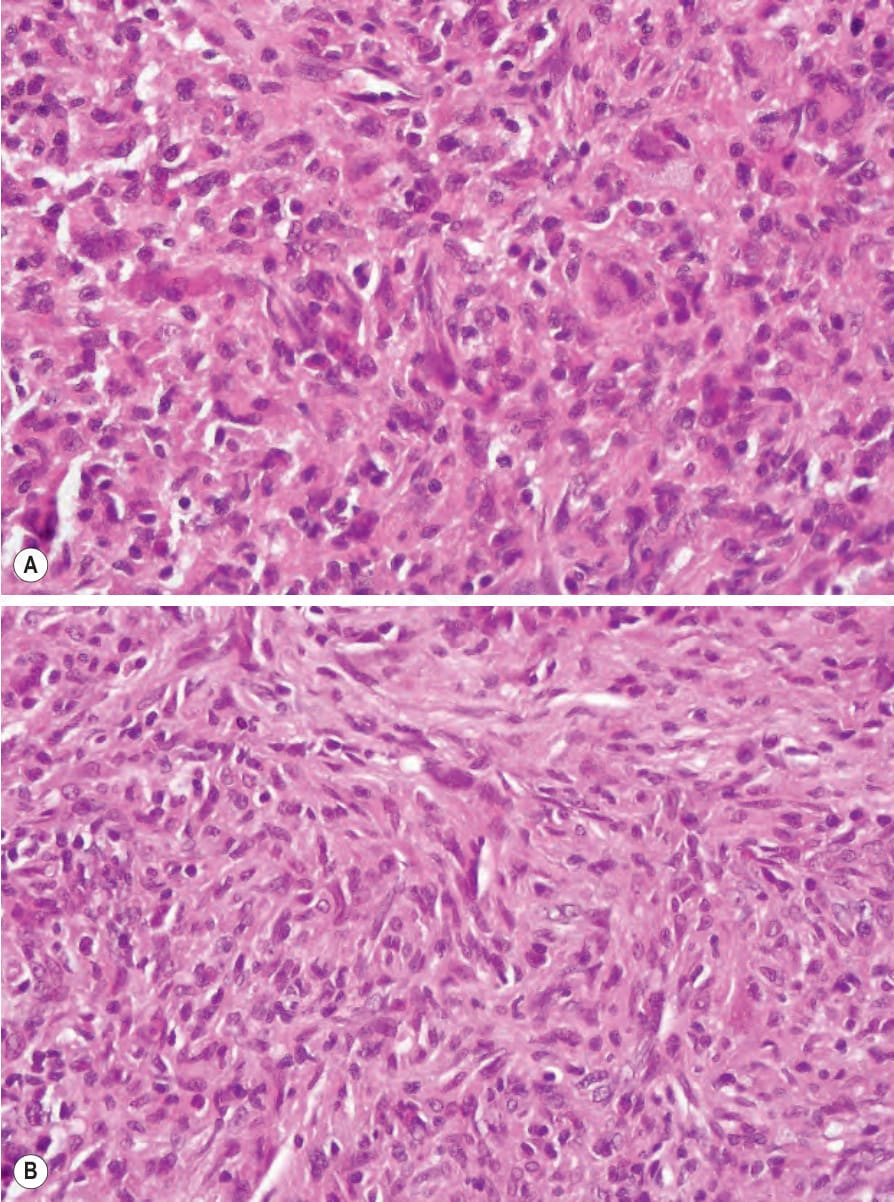
Fig. 29.313 (A, B) Xanthogranuloma: spindle cells predominate in old lesions such that dermatofibroma is often considered in the differential diagnosis.
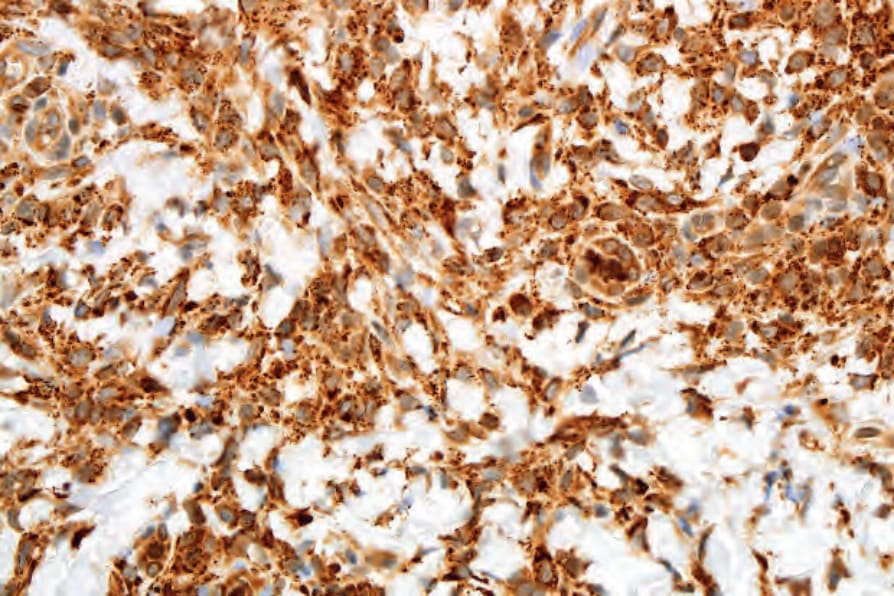
Fig. 29.314 Xanthogranuloma: the histiocytes show strong expression of CD68.
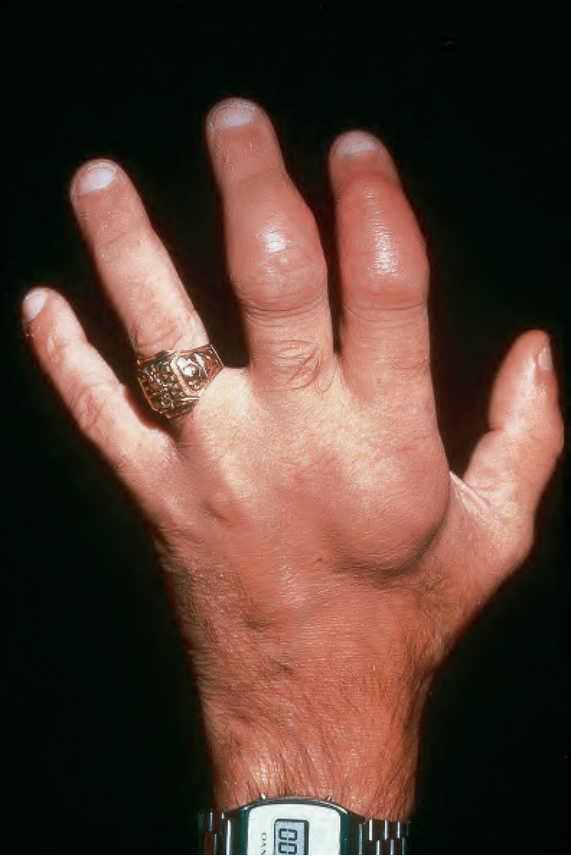
Fig. 29.315 Rosai-Dorfman disease: there is diffuse swelling involving the hand, index, and second fingers. By courtesy of N.S. Pennys, MD, University of Miami School of Medicine, Miami, USA.
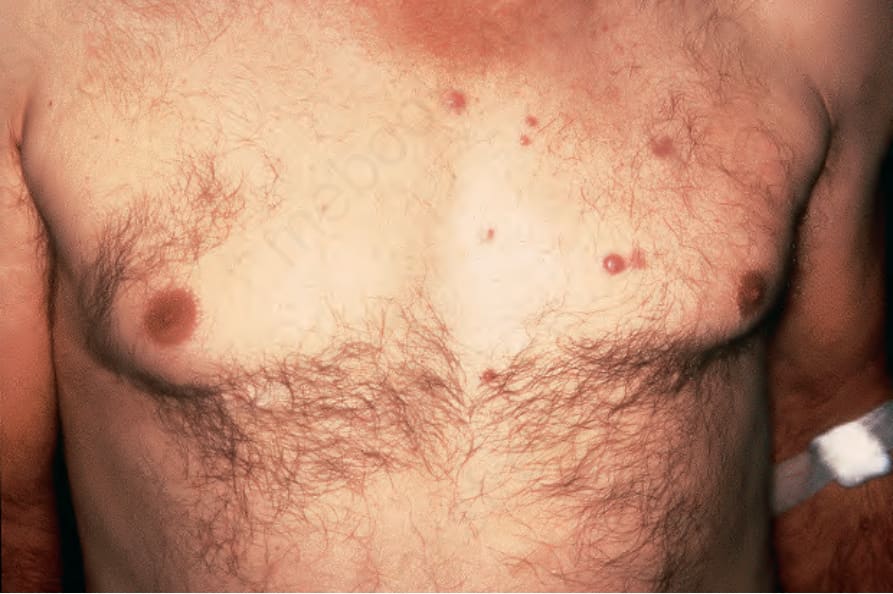
Fig. 29.316 Rosai-Dorfman disease: there are scattered erythematous papules on the left chest. By courtesy of J. Rosai, MD, National Cancer Institute, Milan, Italy.
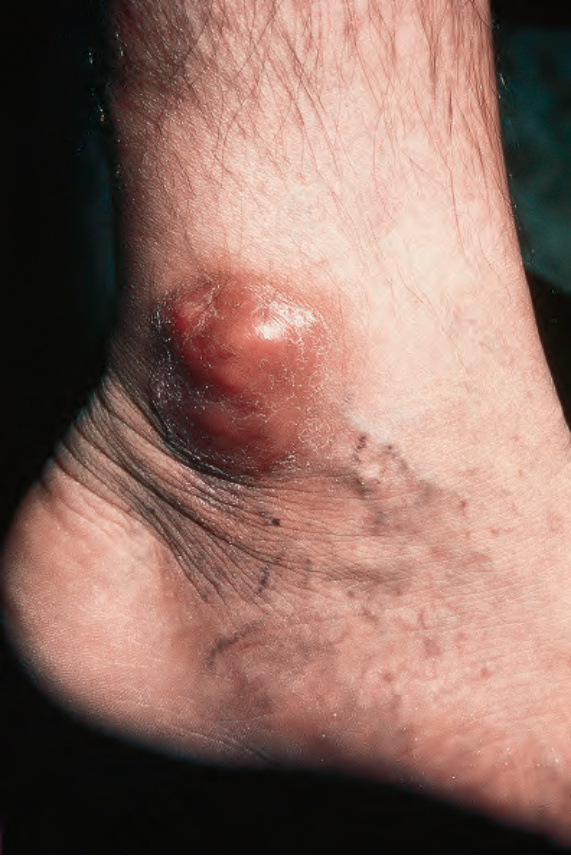
Fig. 29.317 Rosai-Dorfman disease: a brownish-red nodule with scaling overlies the medial malleolus. By courtesy of N.S. Pennys, MD, University of Miami School of Medicine, Miami, USA.
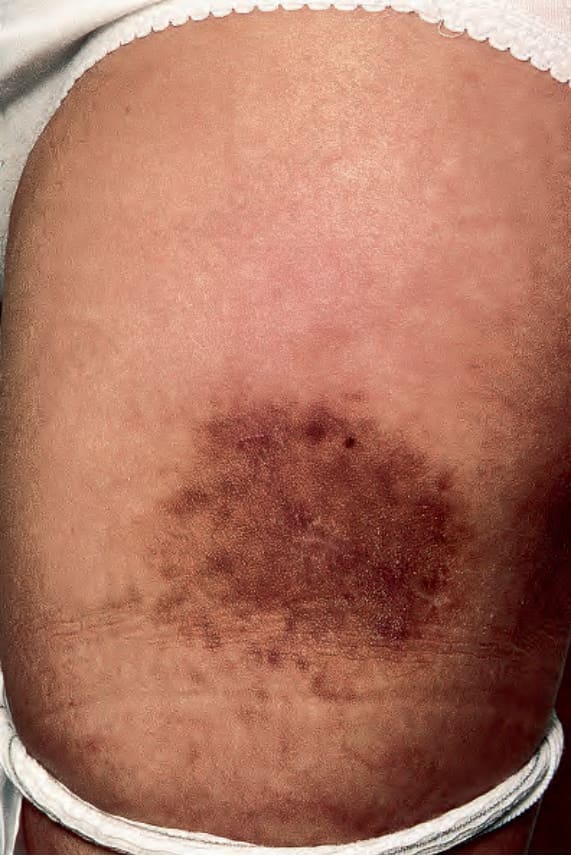
Fig. 29.318 Primary cutaneous Rosai-Dorfman disease: this patient presented with a hyperpigmented indurated plaque. Reproduced with permission from Brenn, T. et al. (2002) American Journal of Dermatopathology, 24, 385–391.
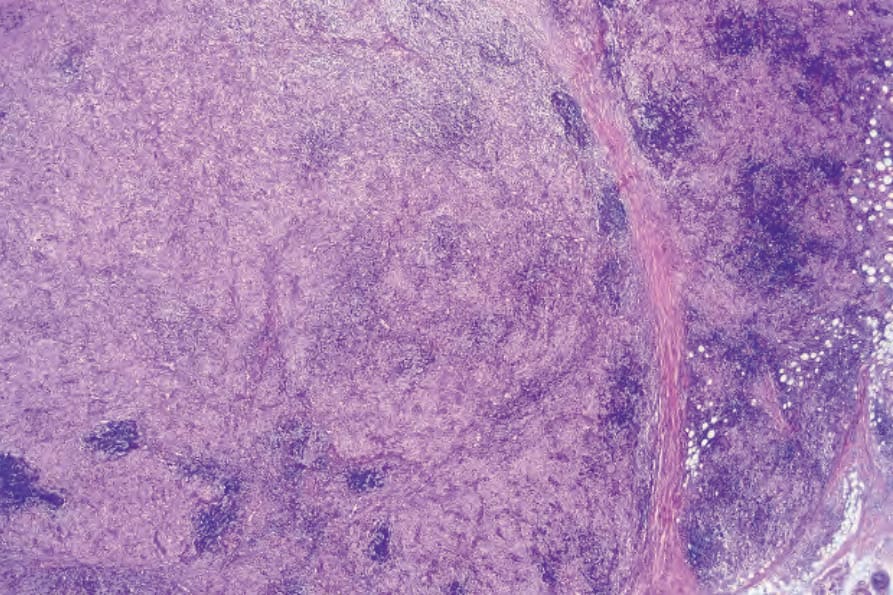
Fig. 29.319 Rosai-Dorfman disease: scanning view showing a dense multinodular cellular infiltrate. Note the lymphoid follicles which are often seen in this condition.
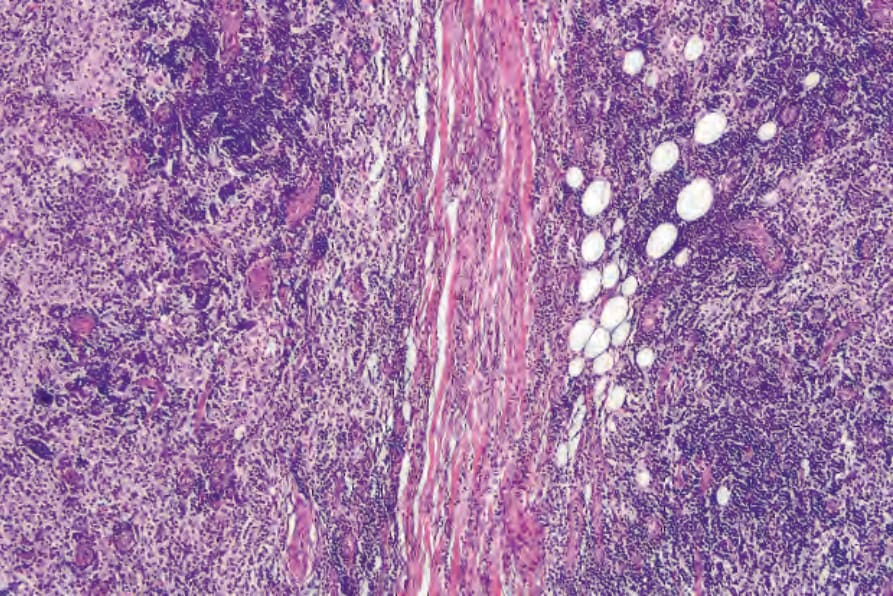
Fig. 29.320 Rosai-Dorfman disease: medium-power view showing two large nodules.
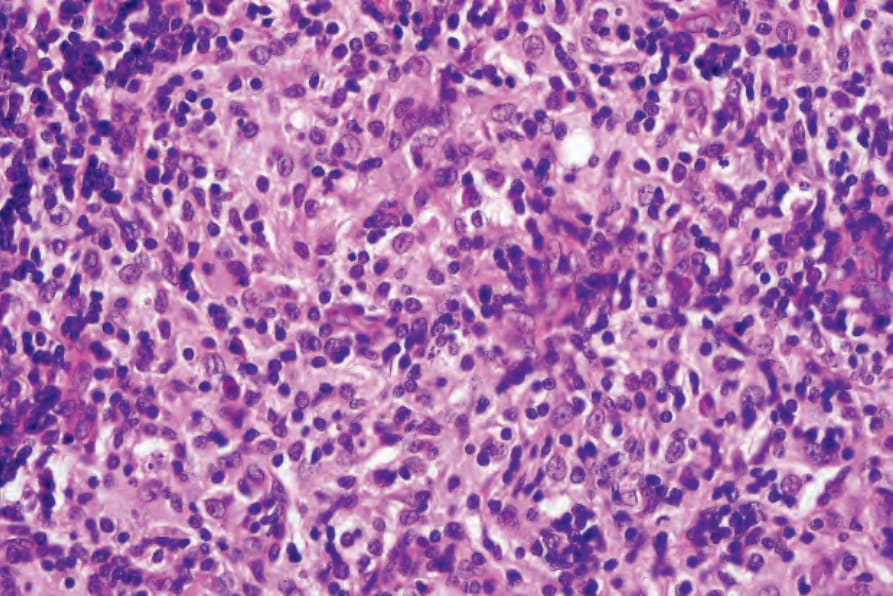
Fig. 29.321 Rosai-Dorfman disease: the infiltrate consists of large histiocytes with abundant eosinophilic cytoplasm admixed with lymphocytes and occasional plasma cells.
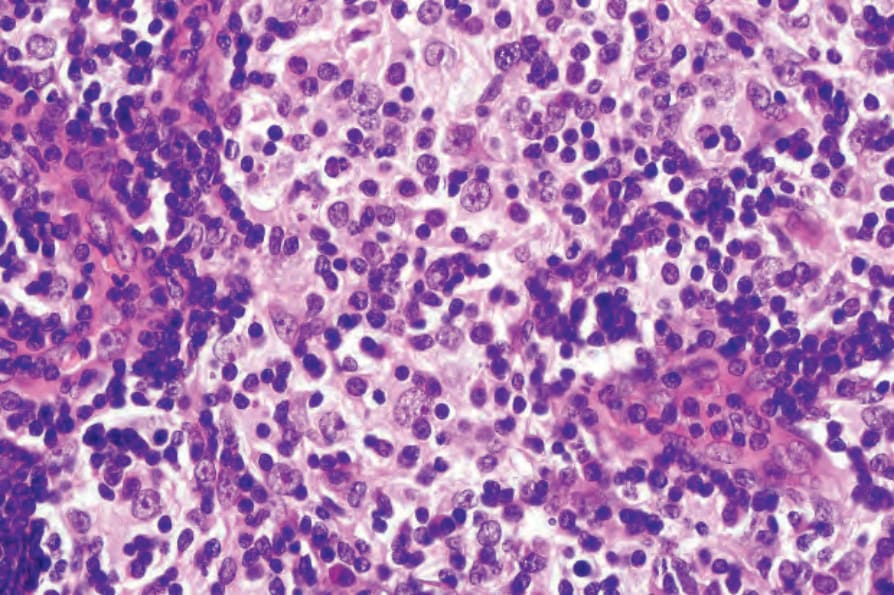
Fig. 29.322 Rosai-Dorfman disease: high-power view showing emperipolesis.
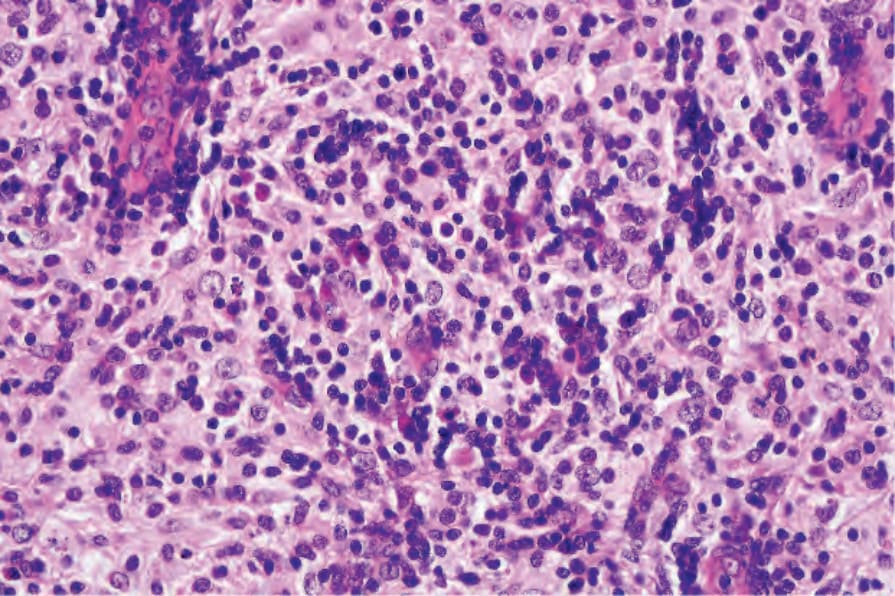
Fig. 29.323 Rosai-Dorfman disease: in this high-power view, there are admixed lymphocytes and plasma cells.
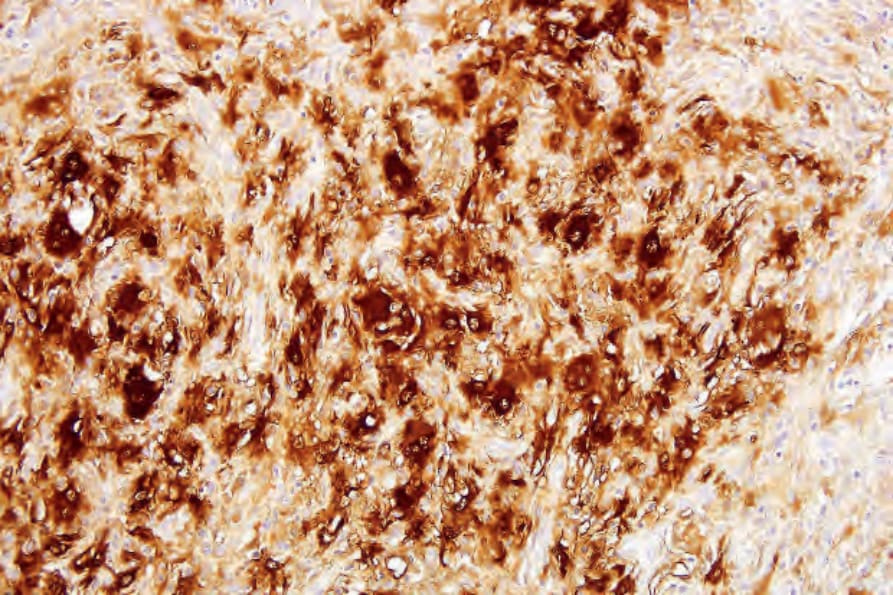
Fig. 29.324 Rosai-Dorfman disease: the histiocytes are S100-protein positive.
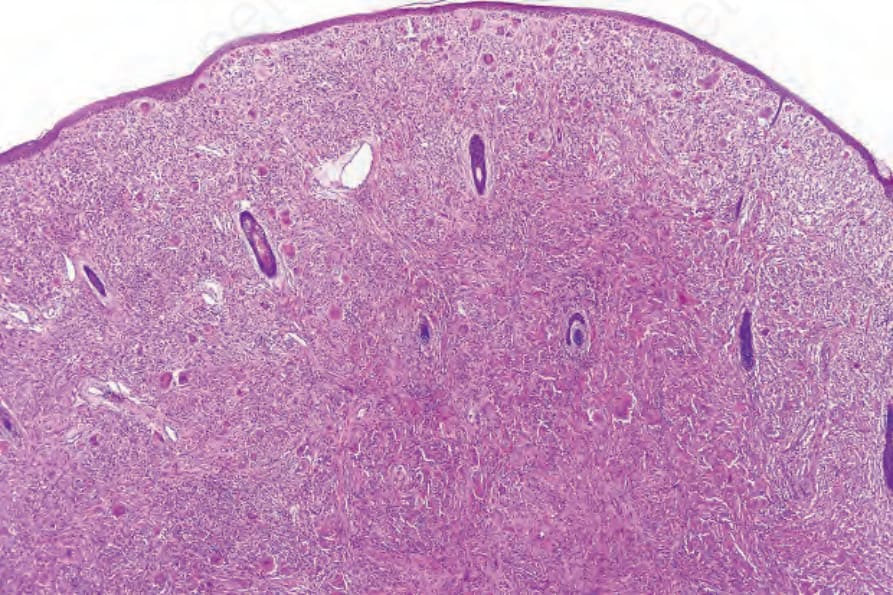
Fig. 29.325 Reticulohistiocytoma: the dermis is expanded by a dome-shaped nodule composed of large histiocytes.
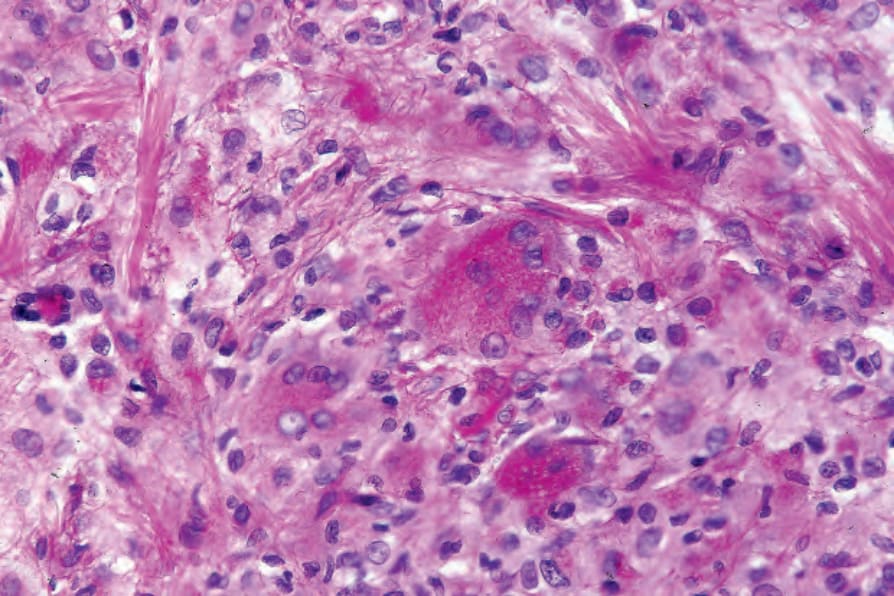
Fig. 29.329 Reticulohistiocytoma: the giant cells are PAS positive, diastase resistant.