Congenital self-healing histiocytosis
Congenital self-healing histiocytosis
Lesions may be solitary or multiple with wide anatomic distribution. Solitary lesions usually present as soft red nodules measuring up to 1 cm in diameter, and may be ulcerated.2,6–8 Multiple lesions are usually red–brown papulonodules ranging from a few millimeters to 1 cm in diameter and tend to appear in successive crops.1,2,7–15 Disease is almost always localized to the skin with no systemic symptoms. Rare cases with visceral involvement and a solitary ocular example have been described.3–6,16 The prognosis is generally good. Most cases undergo complete or partial regression without recurrences, a more aggressive course being rare.3,7
Clinical features Congenital self-healing histiocytosis (CSH) (congenital self-healing reticulohistiocytosis, Hashimoto-Pritzker disease) is a rare condition and is classified as a congenital self-healing variant of LCH.1–9 The sexes are affected equally.
Lesions are present at birth or develop in the perinatal period. They usually consist of widespread erythematous, blue or brown, 2–15 mm diameter macules, papules, and nodules, with sparing of mucous membranes. A solitary red–brown asymptomatic nodule is seen in 30% of cases.10–15 Central ulceration with necrosis and crusting is often present. Vesicular or bullous lesions can also occur.16–20 Healing of the latter may leave anetoderma-like scarring.16 There is predilection for scalp, face, trunk, and proximal extremities, and the palms and soles are sometimes involved.1 A single retinal lesion has been described.21 Very rarely, extracutaneous manifestations including hepatomegaly and mild hematological abnormalities including lymphocytosis occur.5,16 In contrast to LCH, the lesions invariably regress, usually 3–4 months, leaving hypo- or hyperpigmented macules or patches.8 Recurrences are not usually a feature, but patients with CSH require continued observation as there are a small number of patients who go on to develop LCH-like features with recurrent lesions, bone involvement, and diabetes insipidus.22,23 Patients with vesiculobullous lesions may be at particular risk.21
Pathogenesis and histologic features The origin of indeterminate cells is still debated. It has been proposed that they are immature Langerhans precursor cells that have yet to acquire Birbeck granules, or that they are derived from Langerhans cells and have lost Birbeck granules as they migrate toward lymph nodes or that they belong to an independent group of epidermal/dendritic cells.8,9,13,17 Rare cases are association with leukemia or lymphoma, raising the possibility of a common precursor or so-called ‘transdifferentiation’ in some instances.7,13,18,19
Histologically, IDCT is characterized by a dermal infiltrate that may extend into the subcutis, composed of cells that resemble Langerhans cells (Figs 29.288–29.290).4,5 Epidermal involvement is absent.11,12 The constituent cells are usually ovoid or rarely have a more spindle cell morphology.4,5 They possess abundant eosinophilic cytoplasm and oval-to-indented nuclei; nuclear grooves are sometimes seen.4,5,12 Multinucleate giant cells may also be present, as may clusters of lymphocytes, but eosinophils are usually absent.4,5
Histologic features There is an upper dermal infiltrate of epidermotropic large histiocytes with eosinophilic cytoplasm and notched or reniform vesicular nuclei.8,14,24 Neutrophils, eosinophils, lymphocytes, and xanthoma cells are also often present. CSH is histologically indistinguishable from LCH, although it has been reported that a high content of eosinophils, necrosis, and ulceration are more frequently encountered in the former.8 Occasionally, cells with glassy,
Some reports of IDCT describe a predominance of histiocytes with a scalloped or vacuolated appearance and cases with Touton-type giant cells, xanthomatized cells, neutrophils, eosinophils, and plasma cells.1,2,9,10,12,13,19 However, it is possible that this broader morphological spectrum results from inclusion of variants of other forms of non-Langerhans cell histiocytosis, particularly xanthogranuloma, in which there is aberrant S100 protein or CD1a expression.20
The neoplastic cells in IDCT are consistently S100 and CD1a positive but are negative for langerin (Figs 29.291–293).17,19 They lack B- and T-cell markers and are negative for CD30 and FDC markers (CD21, CD23, CD35). Positivity for CD4, CD45, CD68, CD163, factor XIIIA, lysozyme, and HLA-DR is variable, as is the Ki-67 index.3–6,9,13,15,19 One case was clonal with the HUMARA assay.4
1496 Cutaneous lymphoproliferative diseases and related disorders
By definition, no Birbeck granules are seen on electron microscopy, although there is abundant cytoplasm containing lysosomes, phagosomes, and well-developed endoplasmic reticulum.2,7,14
Differential diagnosis IDCT must be distinguished from LCH. Points of distinction include lack of epidermotropism and absence of Birbeck granules. However, electron microscopy may not be available and distinction is not always possible. Differential expression of langerin (CD207) is therefore a useful surrogate for separating these entities.17,19 Uniform expression of CD1a and S100 protein usually allows distinction of IDCT from other forms of non-Langerhans cell histiocytosis.
1497 Xanthogranuloma family
in the periorbital area.3,14–17 These may also occur on the trunk, extremities, scalp, and face.18–22 Lesions may also appear as erythematous to brown patches and plaques on the leg, back and trunk.3
ECD has a varied clinical course. Some patients have few symptoms and an indolent course, whilst others have progressive, potentially lethal disease. CNS involvement is a poor prognostic sign.2,5 There is no recognized treatment, although recent reports suggest that BRAF inhibitors may be very effective in patents harboring the appropriate mutation.23
Pathogenesis and histologic features BRAF mutations are found in around 55% of cases, indicating that this is a truly neoplastic disease.24,25 ECD histiocytes also express a characteristic proinflammatory cytokines and chemokines which are also present in the serum of affected patients, suggesting that the inflammatory milieu plays an important role in the pathogenesis of the clinical manifestations.26,27
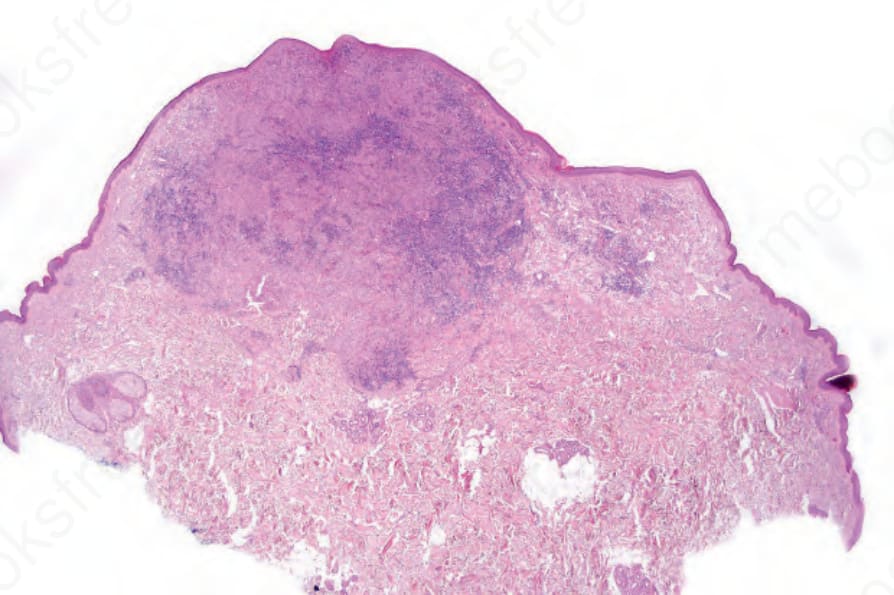
Fig. 29.288 Indeterminate dendritic cell tumor: scanning view of circumscribed dermal tumor nodule.
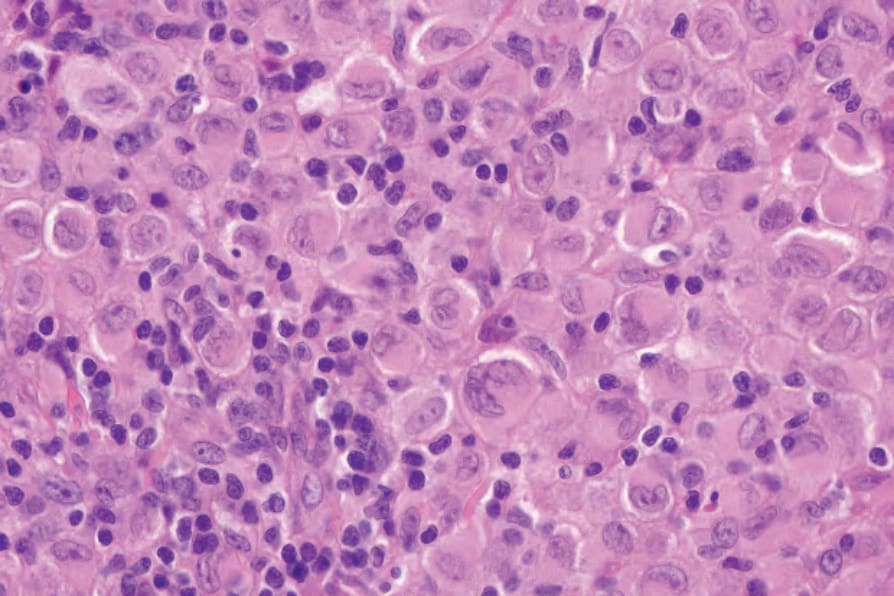
Fig. 29.289 Indeterminate dendritic cell tumor: the appearances are identical to those of Langerhans cell histiocytosis.
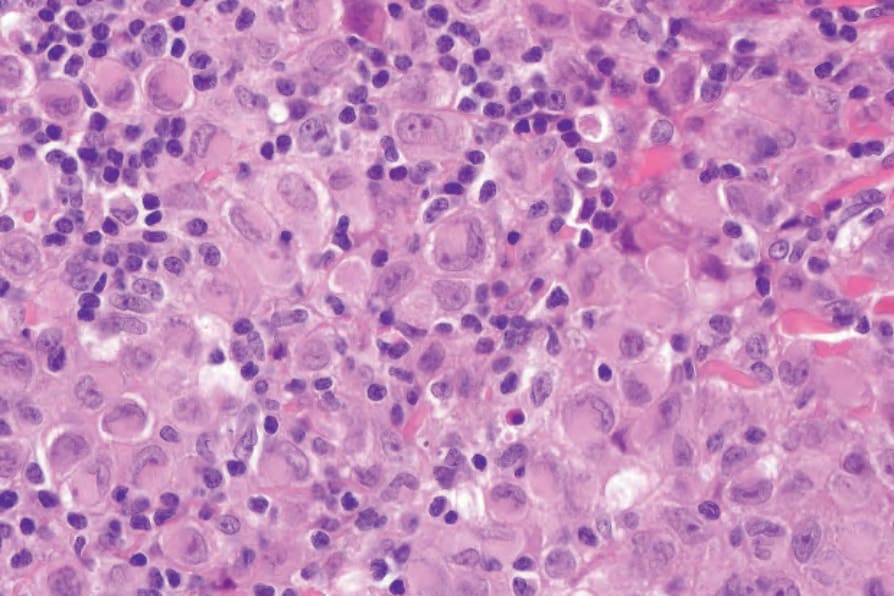
Fig. 29.290 Indeterminate dendritic cell tumor: note the presence of reniform nuclei and delicate nuclear grooves.
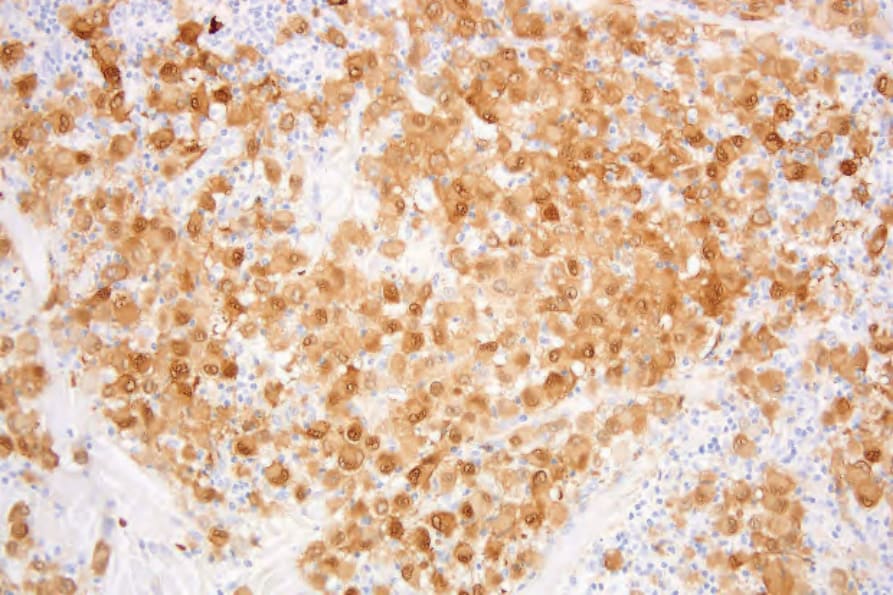
Fig. 29.291 Indeterminate dendritic cell tumor: the tumor cells are S100 positive.

Fig. 29.292 Indeterminate dendritic cell tumor: there is strong expression of CD1a.
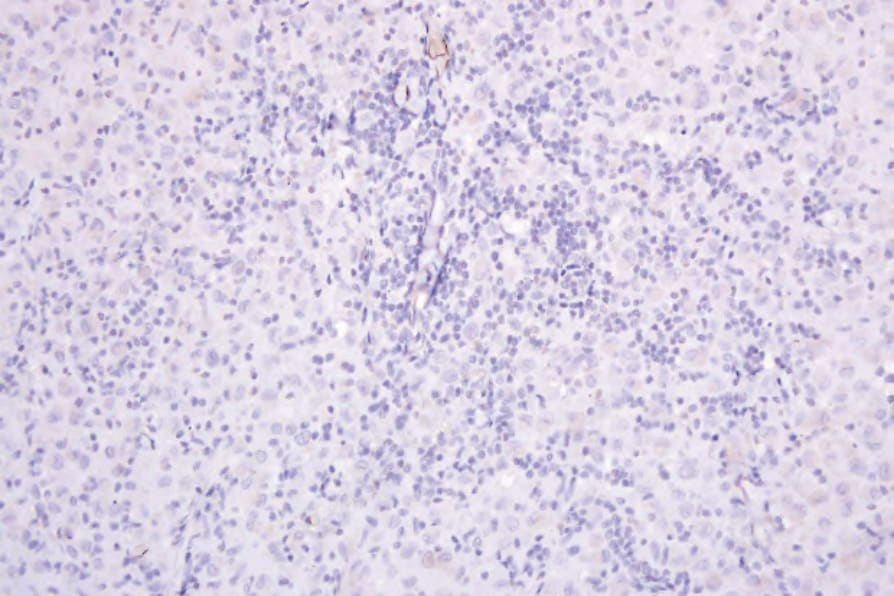
Fig. 29.293 Indeterminate dendritic cell tumor: langerin is negative.