Langerhans cell histiocytosis
Langerhans cell histiocytosis
Clinical features Langerhans cell histiocytosis (LCH) (histiocytosis X, Langerhans cell granulomatosis) is a neoplastic clonal proliferation of Langerhans cells with variable clinical presentation. LCH can be localized to a single site, involve multiple sites within a single system (usually bone), or present as a disseminated multisystem disease.1,2 Previously, and prior to the realization that they represented parts of a spectrum of a single disease process, different synonyms were appended to different clinical presentations. These included eosinophilic granuloma (solitary lesions), Hand-Schüller-Christian disease (multiple lesions), and Letterer-Siwe disease (disseminated disease and/or visceral involvement). Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease) is regarded by some as part of the spectrum of Langerhans cell histiocytoses, but is discussed separately.3–5
LCH is an extremely rare condition with an estimated incidence in children of 1–5 per million of the population per year.6–8 Most cases occur in childhood, although the true incidence in adults is unknown. There is a male predilection.6–9
Patients with unifocal disease (eosinophilic granuloma) are usually older children or adults. Bone and adjacent soft tissue are the most frequently affected sites, particularly the skull, femur, vertebrae, pelvic bones, and ribs. Less commonly, localized disease occurs in lymph nodes, skin, lung, brain, or oral mucous membranes (Fig. 29.269).7,10 Bone lesions are usually lytic with cortical erosions, while at other sites unifocal LCH presents as mass lesions or lymphadenopathy. Cutaneous lesions are deep dermal and/or subcutaneous nodules.8,11,12
Patients with unisystem, multifocal LCH (most examples of Hand- Schüller-Christian disease) are usually children in the first 5 years of life, but the age range is wide (Figs 29.270 and 29.271).13–17 Bone and adjacent
1491 Langerhans cell histiocytosis
marrow, lymph nodes, and lung.7,24 Cutaneous lesions are tiny widespread erythematous, or brownish-red, papules and patches with predilection for the scalp, chest, back, groins, and axillae.27 Perineal and mucosal lesions are common. The rash is often described as eczematoid or seborrheic, although this refers more to distribution than morphology.28 Less commonly, lesions are petichial, purpuric, vesicular, pustular, erosive, or even nodular.8,24,28 In lesions with follicular prominence, the appearances may mimic Darier disease.23,25 Symptoms include fever, weight loss, lymphadenopathy, hepatosplenomegaly, and pancytopenia.8 Radiological studies frequently disclose bone involvement (radiolucent defects), particularly in the skull, ribs, and femur, and miliary or nodular infiltrates in the lungs where, less often, multiple cysts may be found. A hemophagocytic syndrome is a rarely seen in multifocal disease.29 An increased incidence of associated malignancies including acute lymphoblastic leukemia, acute nonlymphoblastic leukemia, HL and NHL, retinoblastoma, medulloblastoma, osteosarcoma, and basal cell carcinoma has been noted, although some of these may be treatment related.30–33
Age is probably not as important as disease extent in determining the prognosis. Patients with unifocal LCH at presentation have a ≥99% survival.1,2,13,14,34 Approximately 10% of patients with multifocal disease die from their illness, while 60% run a chronic course and only 30% achieve complete remission.35,36 Involvement of bone marrow, liver, and lung are reported to be particularly high-risk factors.1,2 Large therapeutic trial suggest that the initial response to chemotherapy is the best predictor of outcome.37–41 Responders have an overall survival of 88–91% compared with 17–34% for nonresponders.37–41 More recently, encouraging results have been obtained using BRAF inhibitors such as vemurafenib.42 Occasionally, patients with unifocal disease subsequently develop multisystem involvement, and even those showing a good response to treatment may suffer from long-term sequelae, including hypothalamic–pituitary dysfunction, cognitive dysfunction, and cerebellar symptoms.1,2,43,44 All patients therefore require long-term follow-up.
soft tissue are most frequently involved, with patients presenting with multiple or sequential destructive bone lesions. The skull and mandible are characteristic sites, and secondary effects may produce the classic triad of osteolytic skull lesions, hypopituitarism-induced diabetes insipidus, and exophthalmos.8,18 Chronic otitis externa is frequent due to involvement of the mastoid or petrous temporal bones.
Multisystem LCH (Letterer-Siwe disease and more extensive presentations of Hand-Schüller-Christian disease) usually presents in infants, and may be present at birth or develop in the neonatal period, although some of these cases may represent examples of congenital self-healing histiocytosis (CSH) (Figs 29.272 and 29.273).7,8,13,19–22 Exceptionally, adults are affected (Fig. 29.274).23–26 There is predilection for skin, bone, liver, spleen, bone
The precise nature of large clusters or sheets of Langerhans cells, seen occasionally in association with some neoplasms or tumorlike conditions, remains to be fully determined, and this includes most cases of isolated LCH of the lung in adults that seems to represent a polyclonal proliferation
1492 Cutaneous lymphoproliferative diseases and related disorders
associated with cigarette smoking.45–48 In some instances, proliferations of LCH may represent a transdifferentiation process. For example, cases of LCH associated with T-lymphoblastic leukemia have been shown to share the same clonal T-cell receptor gene rearrangement, indicative of origin from the same tumor stem cell.49 However, in other instances, LCH-like proliferation associated with various lymphoproliferative disorders appear to represent a localized reactive phenomenon.50
Pathogenesis and histologic features Langerhans cell histiocytosis appears to represent a unique combination of oncogenesis and immune dysregulation.51 Investigations using X-chromosome linked DNA probes or human androgen receptor inactivation (HUMARA) assays have shown the lesional cells to be clonal.44,46,50,52 They also display a less mature phenotype than normal activated Langerhans cells, and show overexpression of various cell cycle related products such as TGF-β receptors I and II, MDM2, p53, p21, p16, Rb, and bcl-2.53–57 The heterogeneous pattern of expression of these factors suggests a degree of regulation, and may explain the slow growth and variable outcome of LCH lesions.53
The local microenvironment also appears to play a role in pathogenesis and the resultant sequelae, such as fever, fibrosis, bone resorption, and necrosis.58,59 Serum cytokine levels, including IL-17A, fms-like tyrosine kinase ligand (FLT3-L), macrophage colony stimulating factor (M-CSF), and vascular endothelial growth factor (VEGF), are elevated in patients with LCH and, in some instances, correlate with extent and severity of disease.60–62 Similarly, high levels of various cytokines have been demonstrated in Langerhans cells, T cells, macrophages, and eosinophils in lesions of LCH including interleukins 1α, 2, 4, 5, 7, 10, and 11, TNF-α, IFN-γ, GM-CSF, and leukemia inhibitory factor (LIF).63 Expression of TNF, IL-11, and LIF has also been shown to correlate with disease severity.63,64
Recurrent BRAF pV600E mutations are present in around 50% of LCH, whilst MAP2K1 mutations are found in a further 19%.65–72 These mutations result in constitutive activation of the MAPK signaling pathway, as do less commonly found mutations in other MPAK pathway genes such as ARAF and MAP3KI.72,73 Mutations affecting other signaling pathways, including PIK3CA, PICK1, and PICK3R2 have also been implicated.70,74
Tumor cells have a rather uniform appearance. They are oval in shape and possess moderately abundant, lightly eosinophilic cytoplasm, and large infolded or reniform vesicular nuclei with thin nuclear membranes and inconspicuous nucleoli. Nuclei often have longitudinal nuclear grooves (coffee bean shape) (Figs 29.275–29.277). Mitotic figures are variable (Fig. 29.278). Neoplastic cells are admixed with variable numbers of eosinophils and, in some cases, with histiocytes (including foam cells and multinucleate forms), neutrophils (often sparse), small lymphocytes, and plasma cells
1493 Langerhans cell histiocytosis
(Fig. 29.279). Eosinophils are numerous in some lesions and may form abscesses with central necrosis rich in Charcot-Leyden crystals. In early lesions, Langerhans cells predominate along with eosinophils and neutrophils, but in later lesions there are increased foamy histiocytes and fibrosis.7
In fully developed cutaneous papules and plaques, the infiltrate is usually dense and bandlike, and may obscure the dermal–epidermal junction. Extravasated red blood cells and epidermotropism with formation of intraepidermal Langerhans cell microabscesses are frequent (Fig. 29.280).8 In some cases, the infiltrate shows a periadnexal, particularly follicular, distribution.25
Lymph node involvement varies from sinusoidal lesions through to partial or complete architectural destruction by tumor cells.7 Exceptionally, a sarcoid-like granulomatous infiltrate has been described.75 In the liver, the infiltrate most often affects the portal tracts and may be associated with bile duct proliferation; a diffuse infiltrate is typically found in the spleen. Bone lesions show a widespread infiltrate, often coupled with fibrosis. Lung lesions comprise a diffuse infiltrate, involving alveoli and alveolar walls, and peribronchial and subpleural deposits. Fibrosis and cysts are common.
The histologic features of focal chronic lesions (eosinophilic granuloma) are similar to those seen in multifocal LCH except that the eosinophils are perhaps present in greater numbers (Figs 29.281–29.283).
1494 Cutaneous lymphoproliferative diseases and related disorders
The hallmark of Langerhans cells is the presence of Birbeck granules, which form in the endosomal recycling area of the cell.76 They are tennis racquet- or rod-shaped organelles measuring 200–400 nm (length) by 33 nm (width), with a zipper-like appearance along the ‘handle’ when viewed by electron microscopy (Figs 29.284 and 29.285). Although specific for Langerhans cells, immunohistochemistry has superseded electron microscopy in confirming the diagnosis of LCH.9
Langerin (CD207) is an antibody to a transmembrane C-type lectin that associates with Birbeck granules, and it is the most sensitive and specific marker for Langerhans cells.53,77,78 Langerhans cells also typically express CD1a, CD4, and S100, and are also positive for vimentin, CD68, and HLA-DR (Figs 29.286 and 29.287).7–9,79 Staining for other B- and T-cell markers, FDC markers (CD21, CD35), and CD30 is negative. The Ki-67 index is highly variable.9
Differential diagnosis Langerhans cell histiocytosis must be distinguished from other histiocytic and dendritic cell neoplasms, including HS and tumors of follicular and IDCs, as well as deposits of acute leukemia. Most cases can be resolved by immunohistochemistry, particularly when antibodies to langerin are available.9,53,59 LCS is differentiated on the basis of overt cytological atypia,
its generally very high mitotic index, and less prominent inflammatory background.7
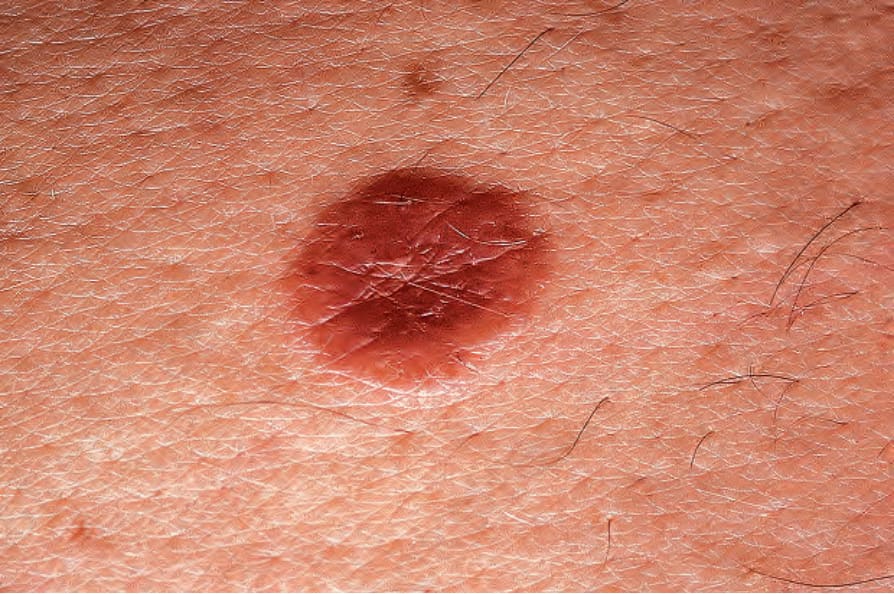
Fig. 29.269 Unifocal LCH (eosinophilic granuloma): this example is a raised erythematous plaque. By courtesy of the Institute of Dermatology, London, UK.

Fig. 29.270 Multifocal chronic LCH: scalp involvement with scale crust and hair loss. By courtesy of the Institute of Dermatology, London, UK.
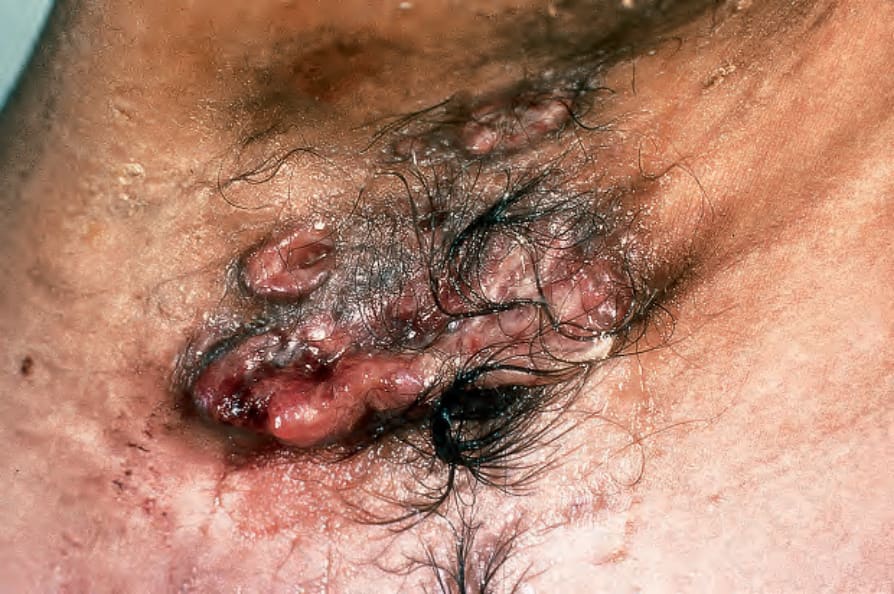
Fig. 29.271 Multifocal chronic LCH: note the vegetative lesions in the axilla. By courtesy of the Institute of Dermatology, London, UK.
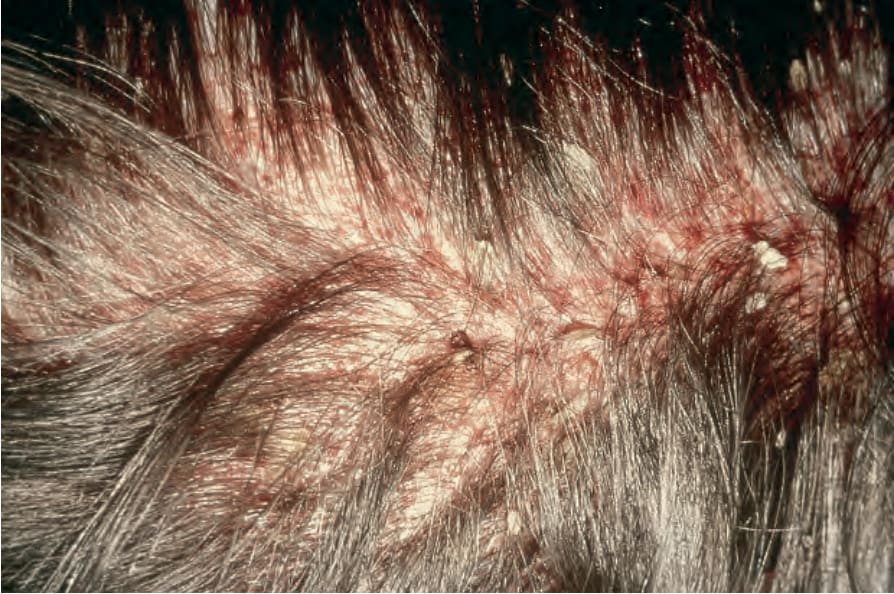
Fig. 29.272 Acute generalized LCH: this scalp shows a characteristic erythematous and scaly eruption. By courtesy of D. Burrows, MD, Royal Victoria Hospital, Belfast, UK.
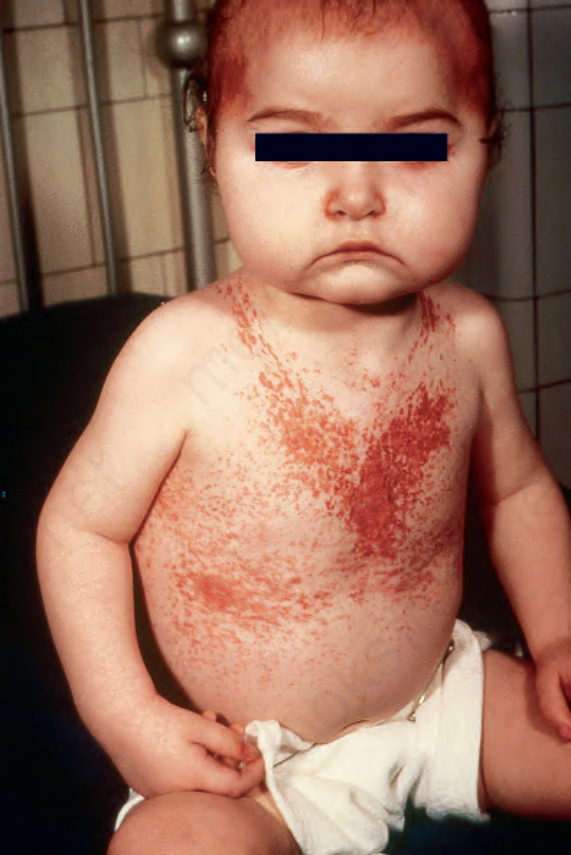
Fig. 29.273 Acute generalized LCH: erythematous eruption showing a characteristic seborrheic distribution. By courtesy of D. Burrows, MD, Royal Victoria Hospital, Belfast, UK.
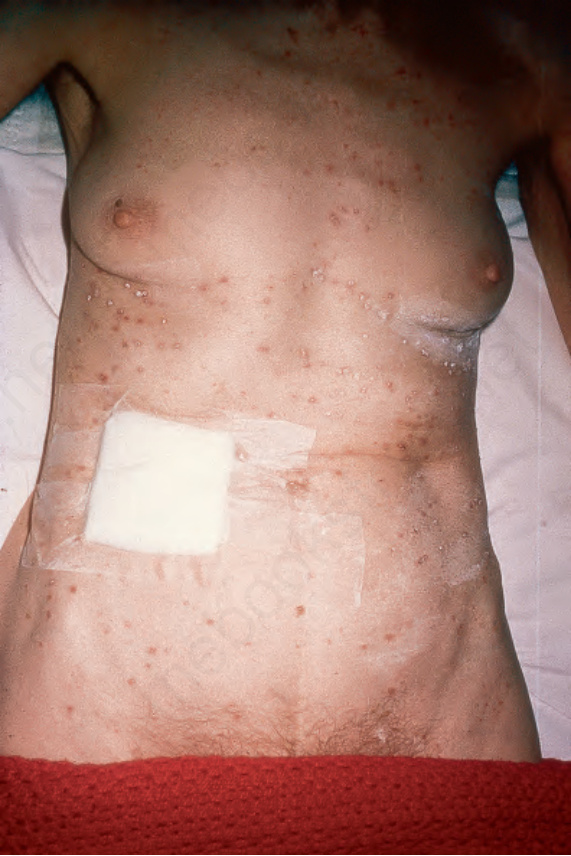
Fig. 29.274 Acute generalized LCH: there are scattered scaly papules over the thorax and abdomen. By courtesy of B. Monk, MD, Kings College Hospital, London, UK.
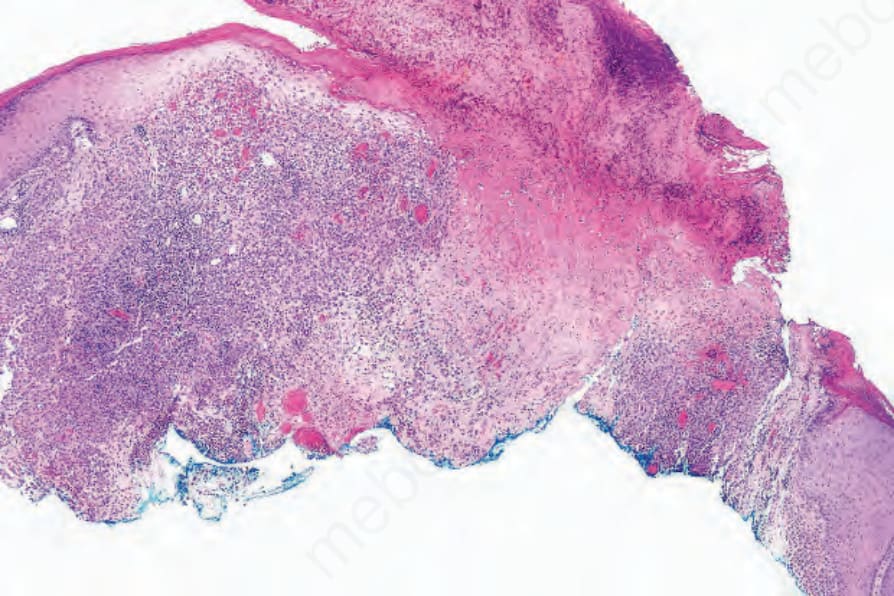
Fig. 29.275 Acute generalized LCH: scanning view of an ulcerated and crusted lesion showing a dense dermal infiltrate.
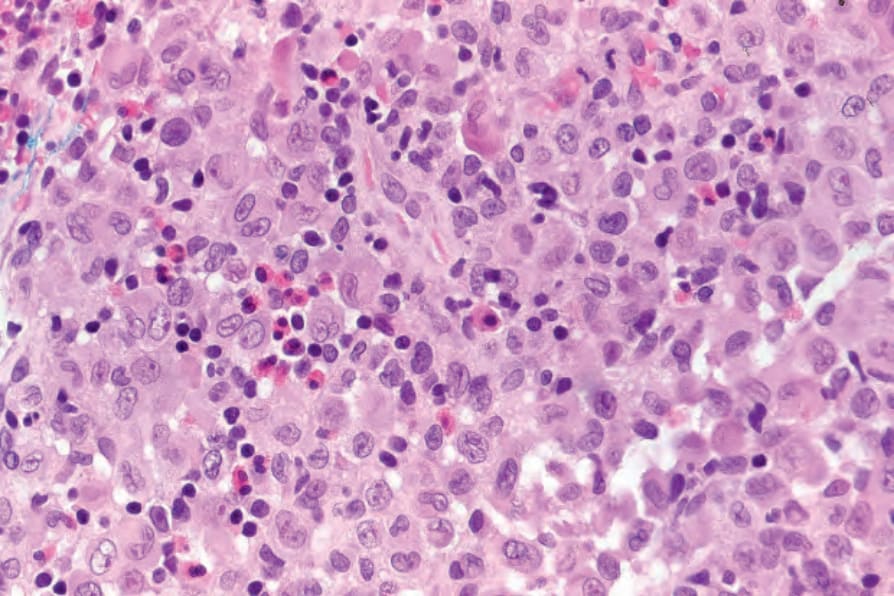
Fig. 29.276 Acute generalized LCH: the infiltrate consists of histiocytes with abundant eosinophilic cytoplasm. Note the lymphocytes and eosinophils.
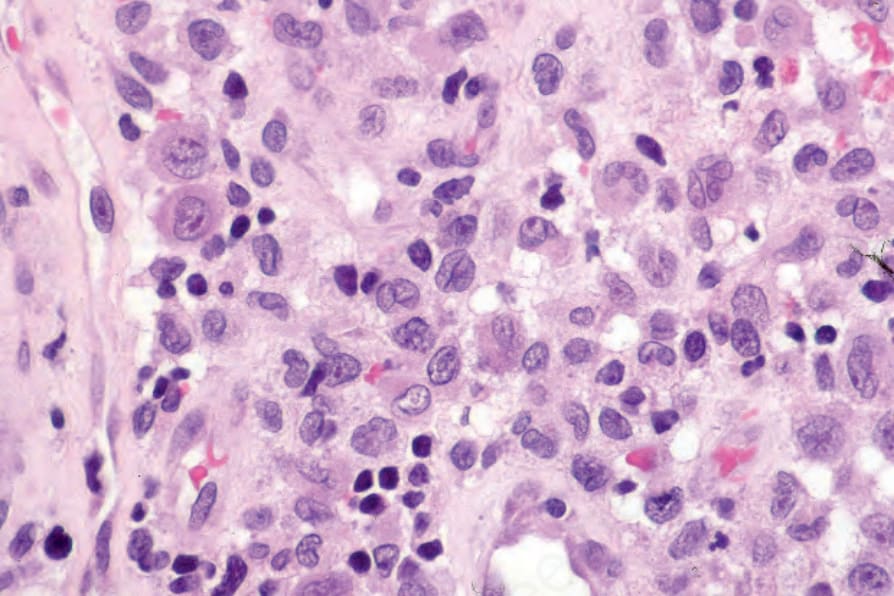
Fig. 29.277 Acute generalized LCH: this field shows typical coffee bean, vesicular nuclei; a nucleus with a longitudinal groove is present in the center of the field.
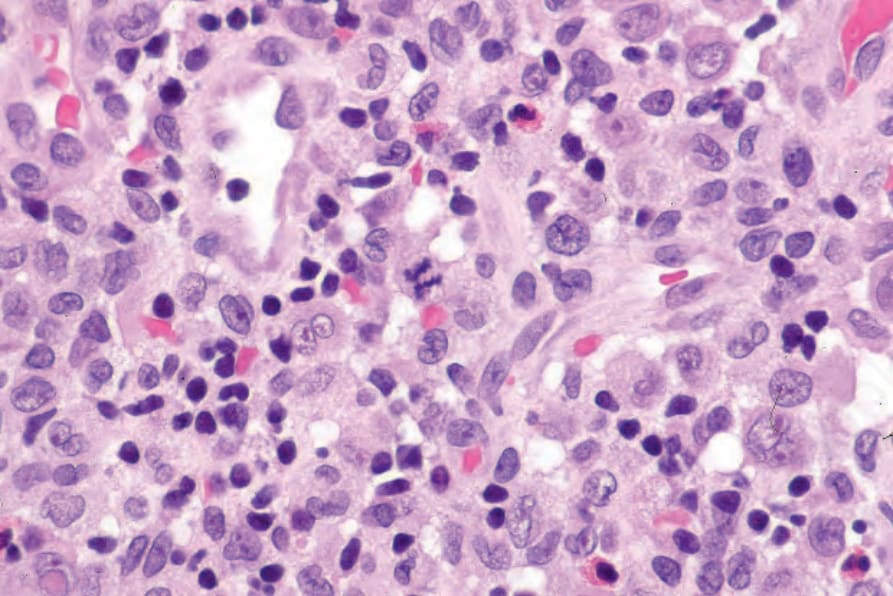
Fig. 29.278 Acute generalized LCH: note the mitotic figure.
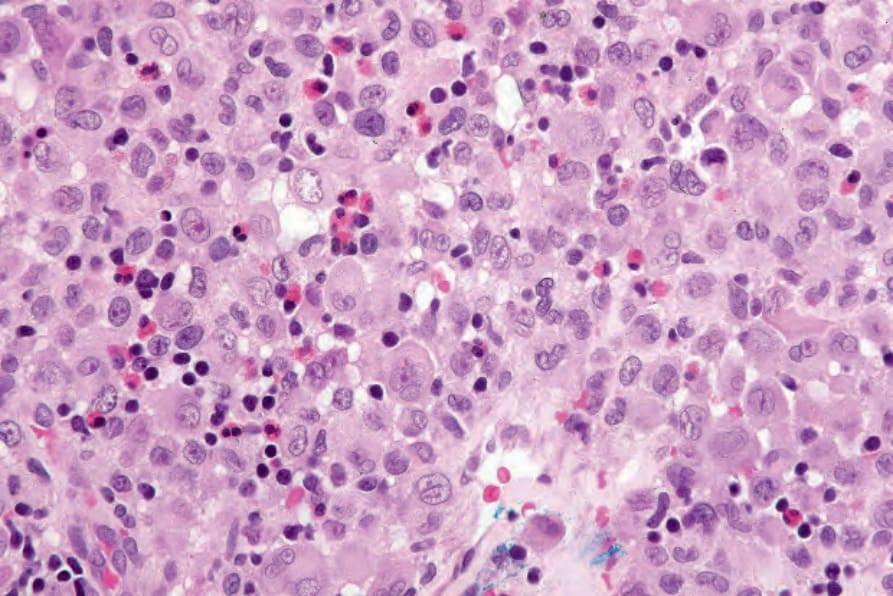
Fig. 29.279 Acute generalized LCH: the conspicuous eosinophils seen here are a variable finding.
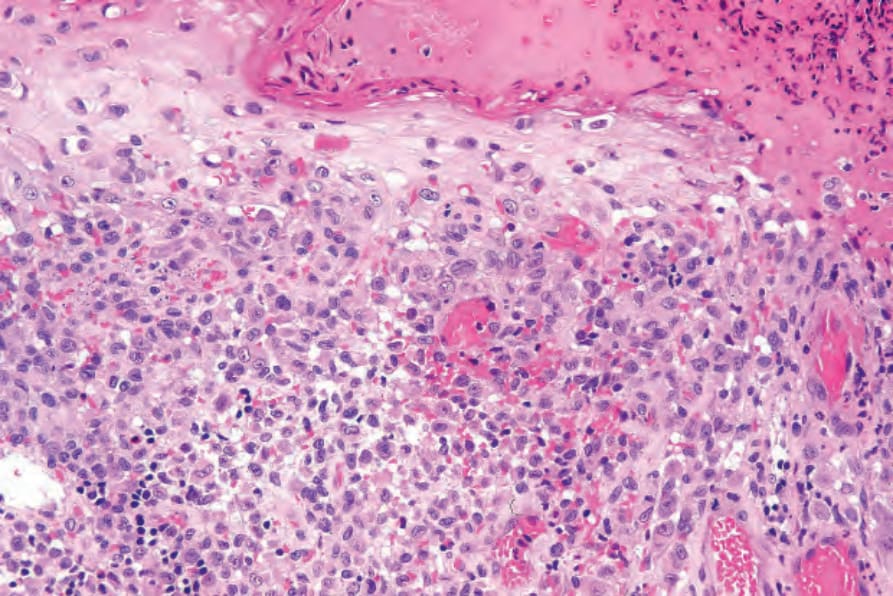
Fig. 29.280 Acute generalized LCH: high-power view showing infiltration of the overlying epidermis, a common feature.
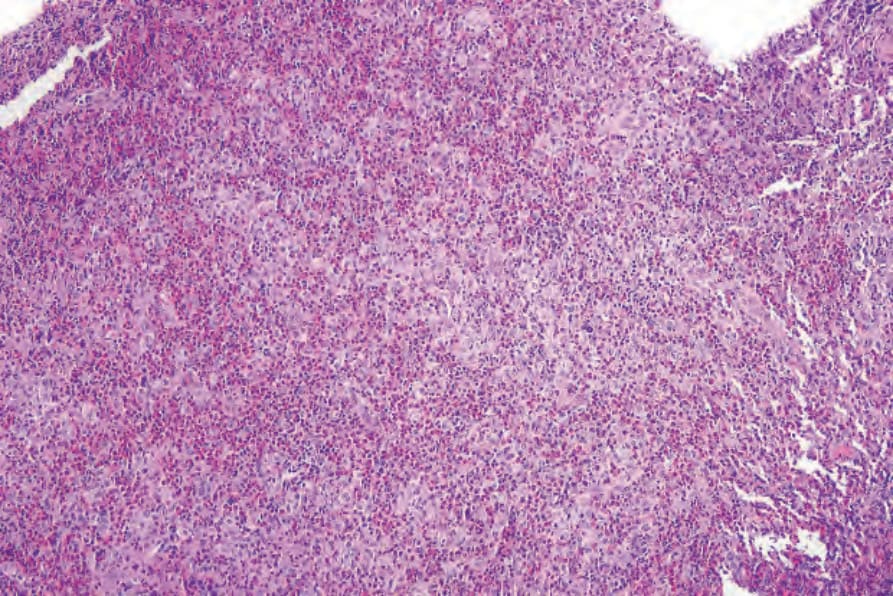
Fig. 29.281 Unifocal LCH (eosinophilic granuloma): low-power view of an oral lesion.
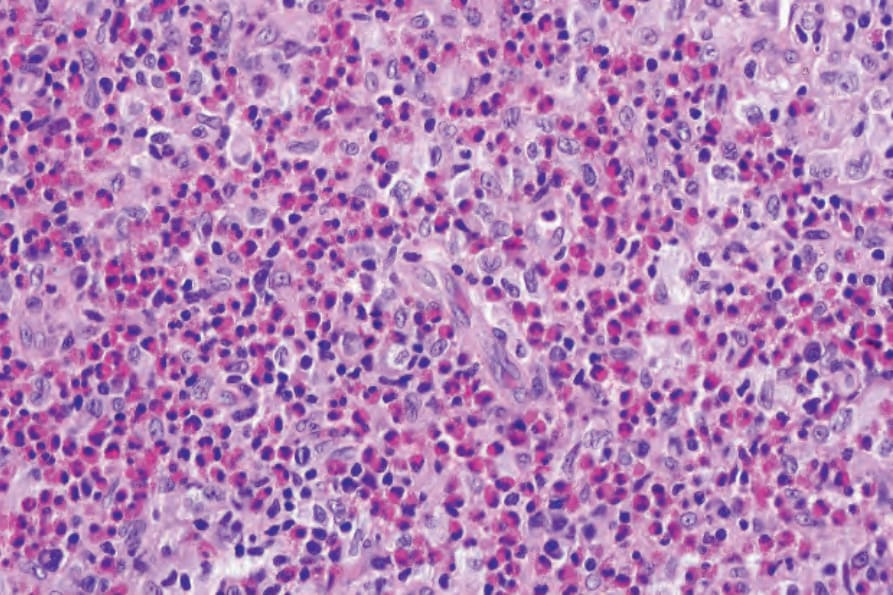
Fig. 29.282 Unifocal LCH (eosinophilic granuloma): note the numerous eosinophils.
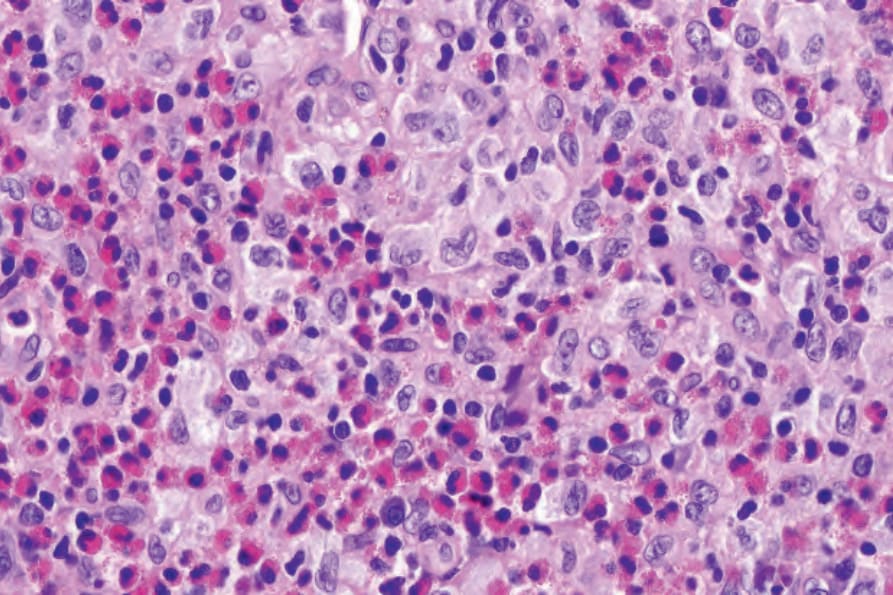
Fig. 29.283 Unifocal LCH (eosinophilic granuloma): high-power view showing typical Langerhans cells.
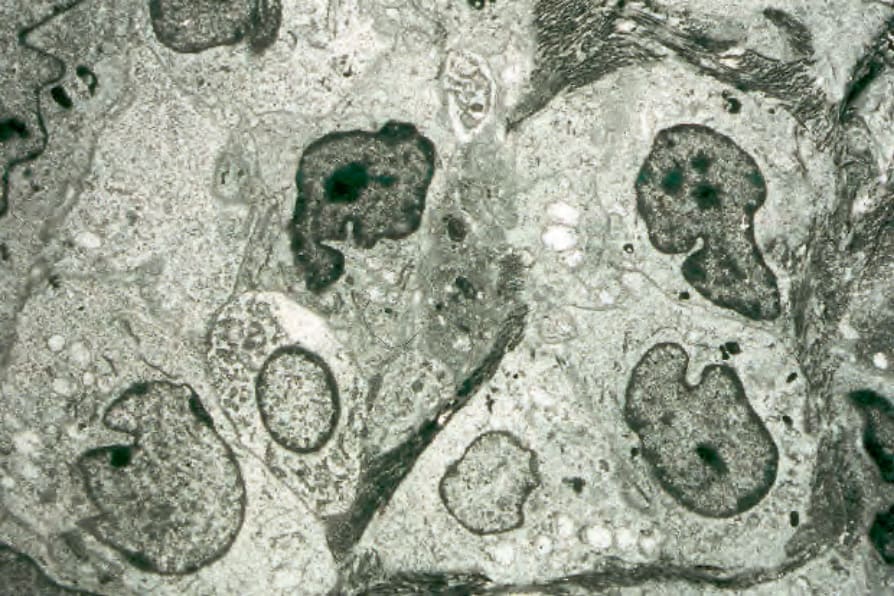
Fig. 29.284 Acute generalized LCH: the infiltrate is composed almost entirely of Langerhans cells. Note the horseshoe-shaped nuclei and abundant cytoplasm.
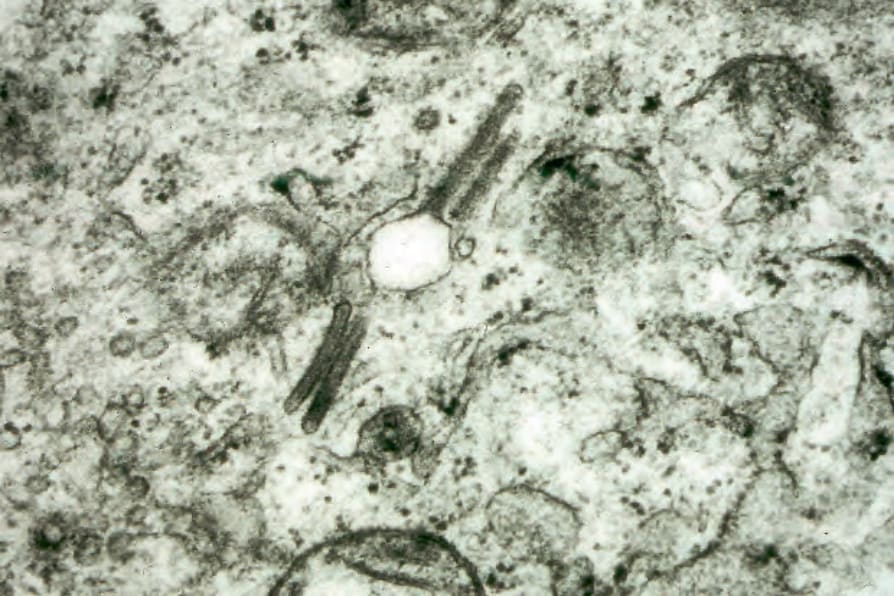
Fig. 29.285 Acute generalized LCH: numerous Birbeck granules are present.
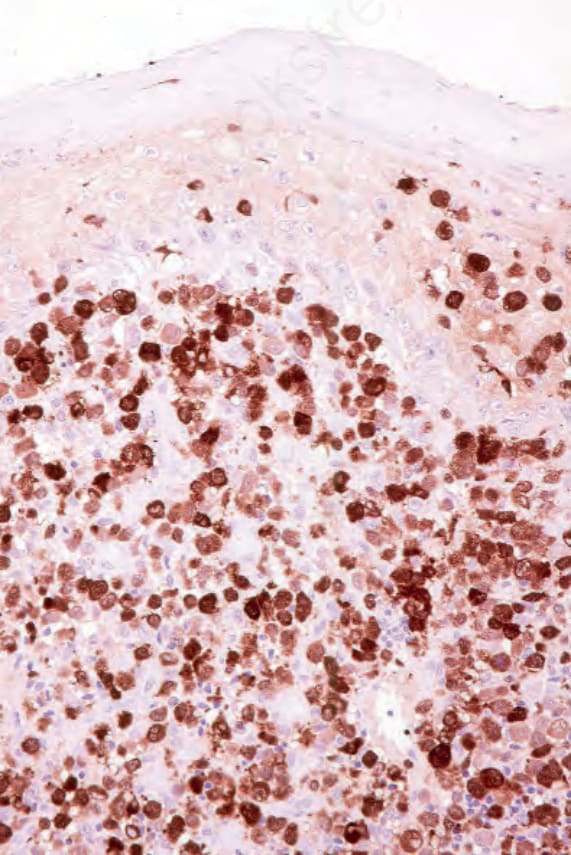
Fig. 29.286 Acute generalized LCH: the Langerhans cells show uniform expression of S100 protein.
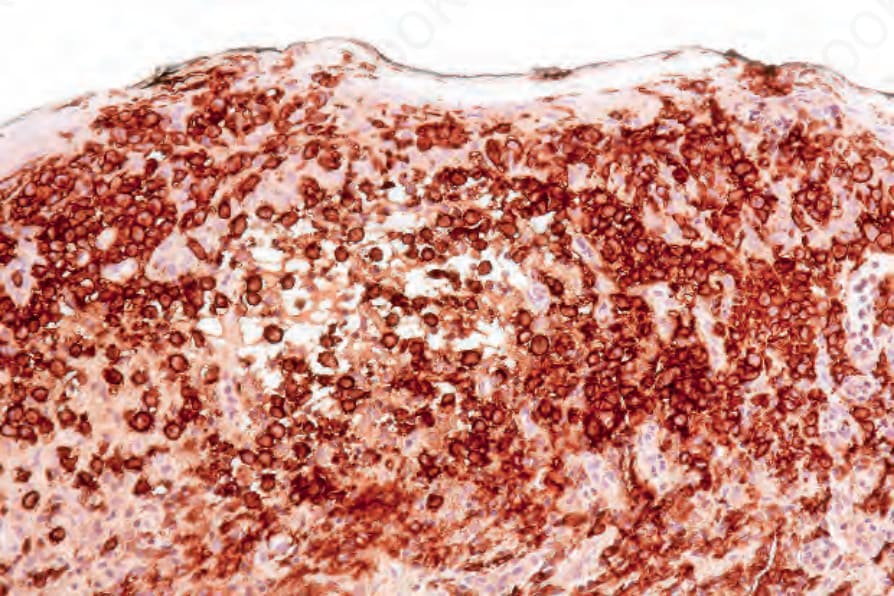
Fig. 29.287 Acute generalized LCH: CD1a is also strongly positive.